Introduction
L'hypertension pulmonaire n'est pas une maladie, mais une situation physiopathologique définie comme une augmentation de la pression pulmonaire moyenne au delà de 25 mmHg, mesurée au cathétérisme cardiaque droit. Les différentes causes d'hypertension pulmonaire sont reprises dans une classification clinique qui comporte 5 groupes, dont l'hypertension artérielle pulmonaire (HTAP), une maladie rare, rapidement progressive et incurable. Le diagnostic différentiel de l'hypertension pulmonaire repose sur un arbre décisionnel établi sur la présence de dyspnée inexpliquée, combinée à une évaluation non invasive comprenant un électrocardiogramme, une radiographie thoracique, des épreuves fonctionnelles respiratoires et une échocardiographie. D'autre examens doivent être réalisés en cas de suspicion de maladie thrombo- embolique pulmonaire, comme la scintigraphie de ventilation/perfusion et l'angioscanner. Seul le cathétérisme cardiaque droit permet de poser avec certitude le diagnostic d'hypertension pulmonaire.
Qu'est ce que l'hypertension pulmonaire?
La circulation pulmonaire normale fonctionne comme un circuit à basse pression (environ 1/6ème de la pressure systémique) avec un double rôle: 1) éviter l'extravasation de liquide vers l'espace alvéolaire et interstitiel; 2) permettre au ventricule droit (VD) de fonctionner à un coût énergétique minimal. Ceci explique qu'il faut imaginer le VD et l'artère pulmonaire (AP) comme une seule entité, dont les fonctions respectives sont décrites comme le couplage ventriculo- artériel.1 L'hypertension pulmonaire (HTP) se défini comme une élévation de la pression artérielle pulmonaire moyenne (PAPm) > 25 mmHg, mesurée au cathétérisme cardiaque droit,2 la valeur normale se situant entre 15 et 20 mmHg. Si l'échocardiographie permet d'estimer la PAP systolique (PAPs), il n'existe pas de définition établie de l'HTP basées sur cette valeur.
L'analyse des déterminants de la PAPm permet de mieux comprendre pourquoi l'HTP n'est pas, en soi, une maladie. En effet, ce qui influence la PAPm apparait en réarrangeant le calcul de la résistance vasculaire pulmonaire (RVP), soit RVP = (PAPm - PAPO) / DC, où PAPO est la pression artérielle pulmonaire d'occlusion (reflétant la pression de l'oreillette gauche, POG) et DC le débit cardiaque.1, 2 L'équation PAPm = (DC x RVP) + PAPO permet immédiatement de comprendre qu'une élévation passive de pression peut être liée à une augmentation de DC (par anémie, thyrotoxicose, shunt artério veineux) ou de pression auriculaire gauche (insuffisance cardiaque, pathologie valvulaire surtout mitrale, fibrillation auriculaire). En revanche, une élévation 'active' de la PAPm par augmentation la RVP peut constituer une situation pathophysiologique plus spécifique, vraisemblablement associée à une vraie modification de la fonction vasculaire pulmonaire et à un remodelage des petites artérioles. Dans ce cas, il est alors légitime de parler de 'maladie vasculaire pulmonaire'.
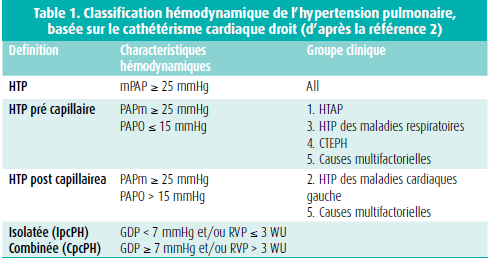
Sur le plan hémodynamique, il est important de distinguer deux présentations particulières, délimitées par une valeur de PAPO: 1) l'HTP pré-capillaire, définie par une PAPO < 15 mmHg; 2) l'HTP postcapillaire (PAPO > 15 mmHg), elle même divisée en HTP post-capillaire isolée (IpcPH), lorsque le gradient diastolique pulmonaire (GDP=PAP diastolique - PAPO) est < 7 mmHg et/ou la RVP est < 3 UW, ou combinée post- et pré-capillaire, lorsque le GDP et > 7 mmHg et/ou la RVP est > 3 UW (table 1).2
La classification clinique de l'HTP permet de distinguer 5 groupes différents, regroupant des pathologies présentant de caractéristiques communes en terme pathophysiologique, clinique et thérapeutiques (table 2).2 En pratique clinique, les formes liées aux maladies cardiaques gauches (groupe 2, en particulier l'insuffisance cardiaque) et celles liées aux maladies respiratoires (groupe 3, principalement dans les pathologies interstitielles) sont de loin les plus fréquentes.2-4 Dans l'immense majorité des cas, elles se présentent comme un symptôme, une anomalie d'un biomarqueur (la PAP) compliquant une affection sous-jacente.2-4 Si elles peuvent intervenir dans le pronostic de la pathologie qui en est la cause, ces formes d'HTP ne font pas l'objet d'un traitement spécifique. En revanche, la présence de signes de souffrance cardiaque droit, suggérant un 'découplage' ventriculo-artériel doit faire l'objet d'une attention particulière et les patients devraient être référés vers des centres experts pour une prise en charge spécifique telle que la transplantation ou l'implantation d'une pompe d'assistance cardiaque.2-4
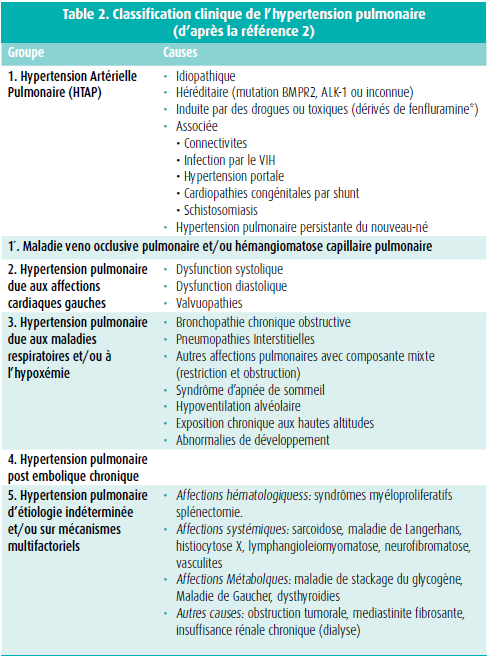
Deux groupes d'HTP représentent des entités pathophysiologiques spécifiques qui doivent être identifiées car elles sont les seules à faire l'objet de stratégies thérapeutiques spécifiques. L'hypertension artérielle pulmonaire (HTAP, groupe 1) est une affection rare (prévalence et incidence minimales respectivement de 15 cas/million et 2,5 cas/million/an),5 grave (médiane de survie sans traitement < 3 ans) et incurable.2 L'HTAP peut se présenter sous forme idiopathique, héréditaire ou suite à une exposition à un toxique (par exemple les dérivés de l'amphétamine ou les traitements par dasatinib). Ces formes représentent environ la moitié des cas qui se présentent dans les centres spécialisés. L'autre moitié des cas d'HTAP se retrouve comme complication d'une pathologie sous-jacente telles que les connectivites (surtout la sclérose systémique), certaines cardiopathies congénitales (conduisant ou non à un syndrome d'Eisenmenger), l'hypertension portale ou encore l'infection par le VIH2. Dans la plupart de ces formes dites associées, l'HTAP est rare: elle complique les cardiopathies congénitales dans 15 % des cas et on la retrouve chez 1 % des patients souffrant de cirrhose et 0,5 % des patients VIH. La sclérose systémique présente la particularité de se compliquer d'HTP dans 8 à 10 % des cas. Cependant, l'HTAP ne représente qu'une faible proportion de patients en comparaison de l'HTP sur pneumopathie interstitielle ou l'HTP sur dysfonction cardiaque gauche.2, 6 Autrement dit, toutes les HTP des connectivites ne sont pas liées à l'HTAP!
L'hypertension pulmonaire post-embolique chronique (CTEPH) est une situation pathophysiologique particulière, pouvant survenir comme complication de 1-4 % des cas d'embolie pulmonaire non résolue.7 Si le facteur déclenchant peut être une obstruction de l'AP par du matériel thrombo-embolique récurrent ou non résolu, la CTEPH est caractérisée par un remodelage vasculaire diffus qui peut être retrouvé dans des zones qui n'ont pas été obstruées initialement. Les facteurs de risque indépendants de la CTEPH sont la splénectomie, les shunts ventriculoartériels, les infections de pacemaker, les affections inflammatoires chroniques ainsi que les antécédents de cancer.7, 8 Il faut aussi tenir compte des troubles de la coagulation qui constituent à la fois des facteurs de risque d'embolie pulmonaire et de CTEPH.7, 8 Il est important de noter que près de 30 % de patients présentant une CTEPH n'ont pas d'histoire de thrombose veineuse profonde et/ou d'embolie pulmonaire documentée.8
Pourquoi le diagnostic différentiel de l'HTP est-il si important?
A travers la classification clinique,2 le clinicien peut comprendre pourquoi il est important de faire la distinction des 5 groupes d'HTP. En effet, il est crucial de distinguer les affections vasculaires pulmonaires des groupes 1 et 4 qui sont associées à un mauvais pronostic sans traitement approprié. Cette règle est également vraie pour les formes graves d'HTP dans les pathologies cardiaques (CpcPH) et pulmonaires (HTP dite sévère lorsque la PAPm est > 35 mmHg) dans la mesure où certains patients sélectionnés peuvent bénéficier d'une prise en charge plus spécifique (voir supra). Il est remarquable de noter que le traitement de l'HTAP et de la CTEPH est déterminé par des recommandations thérapeutiques, fait unique dans les maladies rares. Avec plus de 10 molécules approuvées dans le monde (dont 9 en Belgique), le traitement de l'HTAP est basé sur la combinaison d'antagonistes des récepteurs de l'endothéline (ERA), d'inhibiteurs de la phosphodiéstérase de type 5, de stimulateur de la guanylate cyclase (sGC) et d'analogue de la prostacycline ou de stimulateur des récepteurs de prostacyclines.2 A l'inverse, le traitement de choix de la CTEPH est l'endarterectomie pulmonaire (réalisable dans 60 % des cas) ou, lorsqu'une intervention ne peut être proposée, l'angioplastie par ballon ou un traitement médical par le sGC riociguat.2
Les décisions thérapeutiques dans l'HTAP et la CTEPH doivent impérativement faire l'objet de discussions multidisciplinaires et multiprofessionelles: elles sont la garantie du succès. De plus, les sociétés savantes recommandent formellement que la prise en charge interventionnelle de la CTEPH soit effectuée dans un centre possédant l'expérience de ces procédures à haut risque.2, 7 En Belgique, celles ci sont réalisées à l'UZ Leuven et à l'Hôpital Erasme à Bruxelles. Par ailleurs, la présentation clinique de CTEPH peut être confondue avec celle d'une embolie pulmonaire aigue, ce qui peut conduire à l'initiation de traitements inappropriés.8 Dans cette situation particulière, la présence d'une souffrance VD et d'une élévation de PAPs > 60 mmHg à l'échocardiographie est très en faveur d'une pathologie chronique.
De plus, une identification précoce de l'HTAP dans les groupes à risque permet d'instaurer rapidement un traitement efficace et de ralentir l'évolution clinique, ce qui justifie la mise en place de programmes de dépistage principalement dans la sclérose systémique.9, 10
A l'inverse, la prescription inappropriée de traitements spécifiques de l'HTAP en dehors de leurs indications peut être associée à des effets indésirables. Deux études récentes montrent en effet que l'utilisation de sildenafil dans les HTP d'origine valvulaire9 ou de macitentan dans les CpcPH sur dysfonction ventriculaire gauche10 est associée à un risque plus élevé de détérioration clinique9 ou de rétention hydrosodée10.
Tant le diagnostic différentiel que l'identification précoce des formes rares d'HTP auront des conséquences significatives sur la prise en charge des patients. Dès lors, l'identification de l'origine de l'HTP représente donc un vrai défi pour le clinicien.11
Quelle est la démarche diagnostique de l'HTP?
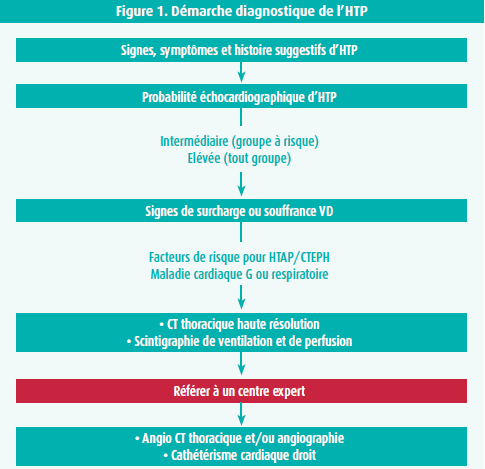
Quelle que soit sa cause, le diagnostic d'HTP ne peut être établi que par une évaluation hémodynamique invasive par cathétérisme cardiaque droit.2 Il est donc important d'identifier les patients pour lesquels ce test est nécessaire. Pour cela, une approche en plusieurs étapes permet de sélectionner les patients pour lesquels un bilan complet sera indiqué.
La suspicion clinique constitue la 1ère étape, très souvent basée sur la présence d'une dyspnée non expliquée. Cependant, les symptômes (dyspnée et fatigue) sont peu spécifiques et les signes (congestion droite) sont souvent tardifs, même dans les formes graves d'http.2, 5-8, 11 Ces signes doivent certainement alerter le clinicien dans les populations à risque d'HTAP: s'il existe une histoire familiale, une connectivité, une HT portale ou une infection par le VIH, la deuxième étape devra être rapidement mise en perspective. De même, la persistance de dyspnée ou de fatigue chez un patient aux antécédents d'embolie pulmonaire et/ou de thrombose veineuse profonde, présentant une thrombophilie ou à risque de CTEPH (cfr supra) aura la même conséquence d'activer la démarche diagnostique.
L'électrocardiogramme, les épreuves fonctionnelles respiratoires, les gaz sanguins et la radiographie de thorax ouvrent la voie vers la 2ème étape: l'échographie cardiaque, le 'maître-des-clés' du diagnostic de l'HTP. La combinaison d'une estimation de la PAPs à partir du jet de régurgitation tricuspide et de signes associés permet d'établir un niveau de probabilité échocardiographique d'HTP. En combinant les données échocardiographiques et les données cliniques (facteurs de risque d'HTAP/CTEPH), il est alors possible de déterminer le niveau de probabilité d'HTP et donc d'identifier les patients chez lesquels un bilan complet sera nécessaire. En pratique, ce dernier sera indiqué chez les patients symptomatiques présentant une probabilité au minimum intermédiaire faisant partie d'un groupe à risque, ainsi que chez les patients présentant une probabilité élevée d'HTP en l'absence de facteurs de risque (table 3). Une fois ces malades identifiés, il est alors recommandé de les adresser à un centre expert pour un bilan complémentaire qui comprendra un cathétérisme cardiaque droit.2

Si l'angiographie pulmonaire par CT s'est imposée comme l'examen de choix dans le diagnostic de l'embolie pulmonaire aiguë, le diagnostic de CTEPH impose la réalisation d'une scintigraphie pulmonaire de ventilation et de perfusion.2, 6 Cette troisième étape est indispensable car ce test révèle la présence d'anomalies de perfusion caractéristiques de CTEPH, l'angio CT (ou l'angiographie pulmonaire) permettant une meilleure localisation des lésions dont l'objectif est de déterminer l'opérabilité de la maladie.2, 6 En d'autres termes, l'angio CT n'est utile dans ce contexte que lorsque la scintigraphie est anormale. Il est recommandé de réaliser l'angiographie pulmonaire dans un centre dont l'expertise dans la prise en charge de l'HTP est confirmée.
Enfin, la dernière étape consiste à réaliser un cathétérisme cardiaque droit, afin de déterminer le type de présentation hémodynamique (table 1) et d'intégrer ces résultats pour poser un diagnostic définitif (table 2). Il est intéressant de noter que le risque de cette procédure est remarquablement faible lorsqu'elle est réalisée dans un centre expert. En effet, une étude multicentrique rétrospective et prospective recent rapporte une mortalité de 0,055 % et une morbidité de 1,1 % dans les centres d'HTP.12
D'autres outils sont indispensables à l'évaluation de la sévérite de l'HTP, en particulier le dosage de peptide natriurétique, l'imagerie par résonance magnétique et les tests fonctionnels comme le test de marche de 6 minutes et l'ergospirométrie.2, 11 S'ils permettent de préciser la sévérité de l'HTP, il ne sont cependant pas suffisamment spécifiques pour en établir le diagnostic.
Que retenir?
Une augmentation de la PAPm, définissant l'hypertension pulmonaire, ne décrit pas une maladie. En revanche, elle identifie une anomalie d'un biomarqueur (la pression) qui peut nécessiter une prise en charge spécifique. Comme toujours en médecine, l'établissement d'un diagnostic précis est la pierre angulaire de toute la prise en charge des formes graves d'HTP, telles que l'HTAP et la CTEPH. Afin d'utiliser au mieux les ressources, de garantir aux patients une prise en charge optimale et d'assurer un contrôle optimal des coûts de la santé, les malades souffrant de formes graves d'HTP doivent être suivi dans un centre spécialisé. L'expérience internationale, mais aussi belge, montre que l'interconnexion et le développement du travail en réseau (couvrant expertise et compétence) permet d'atteindre ces objectifs. C'est ainsi qu'en Belgique, par la qualité de la prise en charge de ces centres, il est possible d'éviter qu'un train en cache un autre…
Références
- Vonk-Noordegraaf, A., Haddad, F., Chin, K. et al. J Am Coll Cardiol, 2013, 62, D22-33.
- Galiè, N., Humbert, M., Vachiéry, J.L. et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J, 2015, 46, 903-975. Eur Heart J, 2016, 37, 67-119.
- Vachiéry, J.L., Adir, Y., Barberà, J.A., et al. Pulmonary hypertension due to heart diseases. J Am Coll Cardiol, 2013, 62, D100-8.
- Seeger, W., Adir, Y., Barberà, J.A. et al. Pulmonary hypertension due to lung diseases. J Am Coll Cardiol, 2013, 62, D109-16.
- Humbert, M., Sitbon, O., Chaouat, A. et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med, 2006, 173, 1023-1030.
- Vachiéry, J.L., Coghlan, G. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev, 2009, 18, 113, 162-169.
- Kim, N., Delcroix, M., Jenkins, na et al.: Chronic thromboembolic pulmonary hypertension, 2013, 62, D92-9.
- Delcroix, M., Kerr, K., Fedullo, P. Chronic Thromboembolic Pulmonary Hypertension: Epidemiology and Risk Factors. Ann Am Thorac Soc, 2016, S3, S201-S206.
- Bermejo, J., Yotti, R., Garcýa-Orta, R. et al. Sildenafil for improving outcomes in patients with corrected valvular heart disease and persistent pulmonary hypertension: a multicenter, double-blind, randomized clinical trial. Eur Heart J, 2017, 0, 1-11.
- Vachiéry, J.-L., Delcroix, M., Al-Hiti, H. et al. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur Respir J, 2018, 51, 1701-886.
- Vachiéry, J.L., Gaine, S. Challenges in the diagnosis and treatment of pulmonary arterial hypertension. Eur Respir Rev, 2012, 21 (126), 313-320.
- Hoeper, M.M., Lee, S.H., Voswinckel, R. et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol, 2006, 48, 2546-2552.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.