Quatre grandes études ont été présentées à la troisième session Hot Line du congrès annuel de la Société Européenne de Cardiologie (ESC) à Munich.
1 L´étude ATTR-ACT
L´étude ATTR-ACT (transthyretine amyloid cardiomyopathie clinical trial) réalisée dans 13 pays montre que la prise de tafamidis améliore la survie ainsi que la qualité de vie chez les patients souffrant d'une cardiomyopathie amyloïde familiale liée à la transthyrétine (CM-TTR).
L'objectif de cette étude était d'évaluer l'efficacité, la sécurité et la tolérance du tafamidis par voie orale à dose de 20 mg ou de 80 mg une fois par jour en comparant avec un placebo pendant une durée de 30 mois chez des patients souffrant d'une CM-TTR.
441 patients ont été randomisés en 3 groupes: un groupe tafamidis 80 mg, un groupe tafamidis 20 mg et un groupe placebo. La randomisation a été réalisée selon un schéma de 2:1:2 de telle sorte que la dose de 80 mg était la plus prescrite (figure 1).
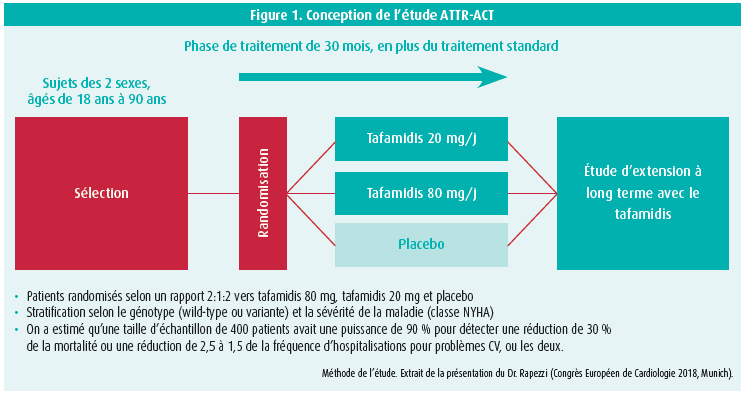
Les critères d'inclusion étaient une preuve par biopsie (cardiaque ou non cardiaque) de la présence de dépôt d'amyloïde, un antécédent d'hospitalisation pour insuffisance cardiaque ayant nécessité de diurétiques, une épaisseur du septum inter-ventriculaire supérieur à 12 mm, un pro-BNP > 600 pg/ ml et une distance de plus de 100 mètres au test de marche. Les critères d'exclusion étaient une classe fonctionnelle NYHA IV, une filtration glomérulaire < 25 ml/min/1,73 m2, la prise d'autres anti-inflammatoires non stéroïdiens ou une denutrition sévère.
Les endpoints combinés primaires d'efficacité étaient toute cause de mortalité ainsi que la fréquence d'hospitalisation due à une insuffisance cardiaque.
Le deuxième endpoint d'efficacité était un changement à 30 mois du test de marche de 6 minutes et de la qualité de vie évaluée par le questionnaire KCCQ-OS (Kansas City Cardiomyopathy Questionnaire).
Finalement, 264 patients ont été randomisés dans le groupe tafamidis et 177 dans le groupe placebo.
L'âge moyen des patients était de 74 ans avec une prédominance masculine (+/- 90 %). La cardiomyopathie TTR de type TTRwt était la plus fréquente (76 %).
La fonction ventriculaire gauche moyenne évaluée à l'échocardiographie était de 48 %. 61 % était en classe fonctionelle NYHA II et 30 % était en classe NYHA III.
Les résultats de survie à 30 mois montrent une diminution significative de la mortalité totale de 30 % dans le groupe tafamidis en comparaison avec le groupe placebo (p = 0,259). Cet effet positif était visible à partir de 18 mois de suivi (figure 2).
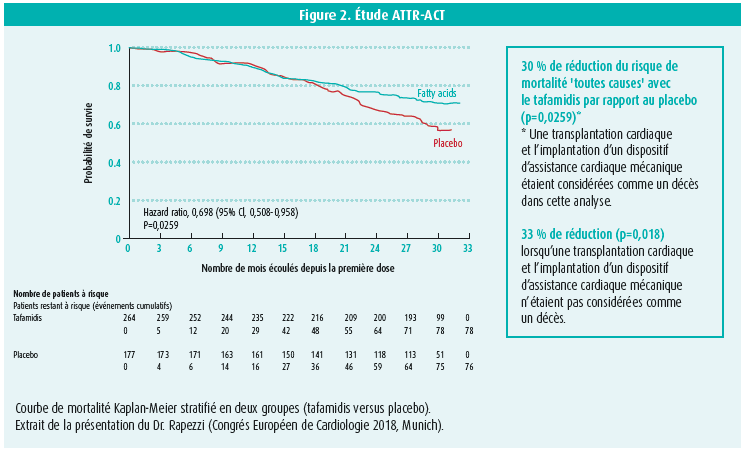
En effet, le nombre de patient décédés après 30 mois de suivi était significativement moins important dans le groupe tafamidis que dans le groupe placebo (186 [70 %] versus 101 [57,1 %], p = 0,006). De plus, le groupe tafamidis présentait significativement moins d'hospitalisations pour insuffisance cardiaque que dans le groupe placebo (138 [2,3 %]) versus 107 [60,5 %], p = < 0,001).
Les résultats du deuxième endpoint montrent également une amélioration significative (p < 0,001) du test de marche de 6 minutes ainsi que de la qualité de vie des patients dans le groupe tafamidis.
Le tafamidis a été très bien toléré et a donc un profil de sécurité rassurant en comparaison avec le placebo chez les patients présentant une CM-TTR.
Le tafamidis semble être ainsi une thérapie efficace pour les patients souffrant de cette pathologie.
2 L'étude COMMANDER HF
L'étude randomisée en double aveugle COMMANDER HF ne montre pas de bénéfice en termes de mortalité ou d'événements cardiovasculaires concernant l'ajout de rivaroxaban à un traitement conventionnelle d'insuffisance cardiaque chez les patients souffrant d'une insuffisance cardiaque à fonction systolique altérée d'origine ischémique en rythme sinusal.
Les critères d'inclusion étaient la présence d'une insuffisance cardiaque chronique à dysfonction systolique avec une fraction d'éjection ventriculaire gauche inférieure à 40 % d'origine ischémique et un antécédent d'hospitalisation pour insuffisance cardiaque dans les 21 jours qui précèdent la randomisation avec un dosage de pro-BNP > 800 pg/ml ou un BNP > 200 pg/ml.
Les patients devaient avoir un traitement optimal d'insuffisance cardiaque et pas d'indication pour une anticoagulation.
Les critères pertinents d'exclusion étaient une fibrillation auriculaire, un risque élevé de saignement, un infarctus aigu du myocarde, une chirurgie cardiaque prévue dans les 28 jours, une maladie valvulaire sévère ou une filtration glomérulaire inférieure à 20 ml/kg/1,73 m2.
Les endpoints combinés primaires d'efficacité étaient toute cause de mortalité, un infarctus du myocarde ou un accident vasculaire aigu.
Les endpoints secondaires d'efficacité étaient la mortalité cardiovasculaire ou une re-hospitalisation pour insuffisance cardiaque ou pour autre événement d'origine cardiovasculaire.
Les principales analyses de sécurité étaient définies soit par une hémorragie fatale, soit par un saignement dans des endroits critiques avec risque potentiel d'handicap permanent.
5 022 patients ont été randomisés en deux groupes: le groupe rivaroxaban (n = 2 507 patients) 2,5 mg deux fois par jour et le groupe placebo (n = 2 515) pendant une durée médiane de 21,1 mois.
L'âge moyen était de 66 ans avec seulement 22 % de femmes. La fraction d'éjection ventriculaire gauche moyenne était de 35 %. La plupart des patients étaient en classe fonctionnelle NYHA II ou III et avaient un traitement optimal.
Contrairement à ce qu'espéraient les auteurs, les résultats cliniques étaient défavorables. En effet, il n'y avait pas de différence significative entre les deux groupes tant pour l'endpoint primaire (p = 0,27) que secondaire.
Concernant les études de sécurité, il n'y avait également pas de différence entre les deux groupes concernant les hémorragies fatales ou à des endroits critiques avec risque potentiel d'handicap permanent reflétant un profil de sécurité tout à fait rassurant.
3 L'étude MITRA-FR
L'étude MITRA-FR (Percutaneous Repair With the MitraClip Device for Severe Functional/Secondary Mitral Regurgitation) montre que la réparation percutanée d'une d'insuffisance mitrale sévère d'origine fonctionnelle par MitralClip ne diminue pas la mortalité ni les hospitalisations pour insuffisance cardiaque.
Les critères d'inclusion étaient une insuffisance mitrale sévère fonctionnelle (orifice régurgitant > 20 mm2 ou un volume régurgité > 30 ml) symptomatique malgré un traitement médicamenteux optimal conduit. Le patient devait également avoir un antécédent d'hospitalisation pour insuffisance cardiaque dans les 12 mois qui précédaient la randomisation, une fraction d'éjection entre 15 % et 40 % et ne pas être éligible pour une réparation chirurgicale conventionnelle.
Les endpoints primaires étaient toute cause de mortalité ou une hospitalisation pour insuffisance cardiaque à 12 mois.
307 patients ont été randomisés en deux groupes: 152 patients dans le groupe MitralClip et 152 dans le groupe contrôle (3 patients étaient exclus pour un problème de consentement.).
Les deux groupes présentaient des caractéristiques à l'inclusion similaire. L'âge moyen était de 70 ans avec une prédominance masculine (78 %). 62 % des patients souffraient d'une cardiopathie ischémique et deux tiers étaient en classe NYHA III-IV. La fraction d'éjection ventriculaire gauche moyenne était de 33 %.
Les résultats de sécurité montrent que la technique est sûre et efficace (aucun décès ni de conversion en chirurgie à coeur ouvert pendant l'étude) avec 96 % de succès d'implantation.
Les résultats en termes de mortalité ou de re-hospitalisation pour insuffisance cardiaque sont décevants. En effet, il n'y avait pas de différence significative entre les deux groupes (83 (54,6 % versus 78 (51,3 %) p = 0,53).
Soulignons tout de même, le manque de puissance potentielle statistique vu le faible échantillon. Dès lors d'autres études randomisées avec une plus grande population sont nécessaires.
4 L'étude GLOBAL LEADERS
L'étude GLOBAL LEADERS montre qu'un traitement antiplaquettaire incluant une combinaison d'acide acétylsalicylique et de ticagrelor pendant un mois suivi de 23 mois de ticagrelor seul n'est pas supérieur en termes de mortalité ou de nouveaux infarctus aux traitements antiplaquettaires standards.
Ils ont comparé une stratégie expérimentale, définie comme une combinaison d'acide acétylsalicylique et de ticagrelor pendant un mois, suivi de 23 mois de ticagrelor seul chez des patients ayant soit présenté un infarctus aigu du myocarde, soit une maladie coronaire stable aux stratégies de référence. Celles-ci étaient définis soit comme une combinaison d'acide acétylsalicylique et de ticagrelor pendant un an, suivi de 12 mois d'acide acétylsalicylique seul chez les patients ayant présenté un infarctus aigu du myocarde, soit une combinaison d'acide acétylsalicylique et de clopidogrel pendant 12 mois, suivi de 12 mois d'acide acétylsalicylique seul chez les patients présentant une maladie coronaire stable.
15 991 patients ont été randomisés 1:1 en deux groupes dans 130 centres dans le monde.
Les endpoints primaires étaient toute cause de mortalité ou l'apparition d'une onde Q reflétant une nouvelle séquelle d'infarctus du myocarde pendant un suivi de 2 ans.
Après l'exclusion de 31 patients, 7 980 patients se sont retrouvés dans le groupe stratégie expérimentale et 7 988 dans le groupe stratégie standard.
Les caractéristiques de base entre les deux groupes étaient similaires. L'âge moyen était de 64 ans incluant 23 % de femme et 75 % avait une maladie coronaire d'un vaisseau.
En termes de présentation clinique, 53 % avait une maladie coronaire stable et 47 % avait présenté un infarctus du myocarde. Les résultats concernant la sécurité sont tout à fait rassurants. Il n'y a aucune différence significative entre les deux groupes en terme de sécurité.
à un an de suivi, les résultats montrent une diminution significative de toute cause de mortalité et/ou de l'apparition de nouvelles ondes Q dans le groupe expérimental (156 [1,95 %] versus 197 [2,47 %] de décès respectivement p = 0,028).
Par contre, à 2 ans de suivi on ne retrouve plus de diminution significative de la mortalité ou de l'apparition de nouvelles ondes Q (304 [3,81 %] versus 347 [4,37 %] respectivement, p = 0,073) (figure 3).
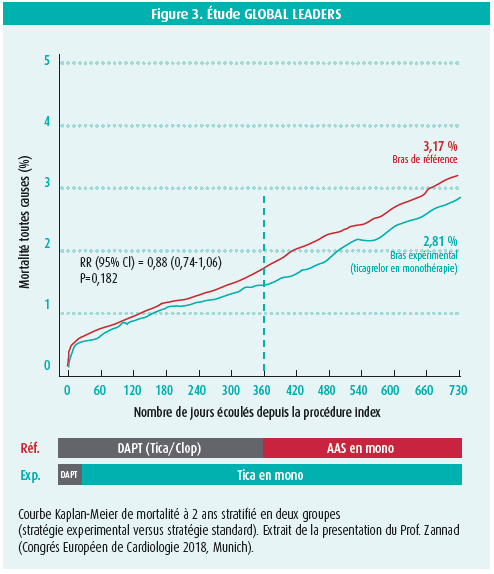
En conclusion, il n'y aurait pas de bénéfice de continuer le ticagrelor après 12 mois de traitement.
Notons tout de même, une moins bonne compliance médicamenteuse dans le groupe expérimental que dans le groupe standard.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.