Pulmonale arteriële hypertensie: een zeldzame ziekte
Pulmonale hypertensie is geen ziekte, maar een pathofysiologische situatie die wordt gekenmerkt door een abnormale stijging van een biomarker, in casu de druk in de longslagader (PAPm), tot meer dan 25 mmHg gemeten tijdens een rechterhartkatheterisatie. 1
Naargelang van de wigdruk (pulmonale arteriële occlusiedruk) onderscheiden we twee hemodynamische situaties:
- precapillaire pulmonale hypertensie: wigdruk < 15 mmHg;
- postcapillaire pulmonale hypertensie (wigdruk > 15 mmHg); wordt verder ingedeeld in geïsoleerde postcapillaire hypertensie, als de pulmonale diastolische gradiënt (diastolische PAP - wigdruk) < 7 mmHg is en/of de pulmonale vaatweerstand ≤ 3 WE, en gecombineerde pre- en postcapillaire pulmonale hypertensie, als de pulmonale diastolische gradiënt ≥ 7 mmHg is en/ of pulmonale vaatweerstand > 3 WE.1, 2
De klinische classificatie van pulmonale hypertensie telt vijf groepen, waarin de ziektes worden gebundeld op grond van gemeenschappelijke pathofysiologische, klinische en therapeutische kenmerken (tabel 1).2 Veruit de frequentste zijn pulmonale hypertensie als gevolg van een ziekte van het linkerhart (groep 2, vooral hartfalen) en pulmonale hypertensie als gevolg van een ademhalingsziekte (groep 3, hoofdzakelijk interstitieel longlijden).1-3
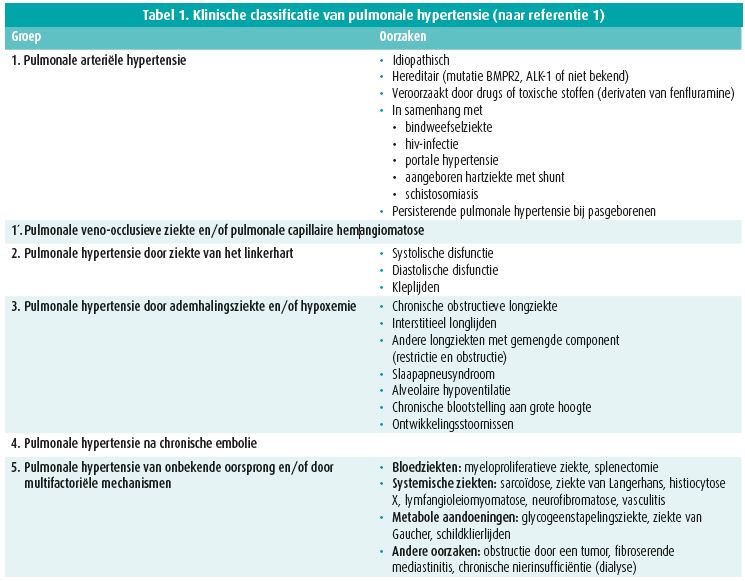
Pulmonale arteriële hypertensie (groep 1) en pulmonale hypertensie na chronische embolie (CTEPH, groep 4) zijn specifieke pathofysiologische entiteiten die gediagnosticeerd moeten worden, aangezien dat de enige zijn waarvoor een specifieke behandeling bestaat.1 In die twee domeinen is de laatste 10 jaar veel vooruitgang geboekt en is het therapeutische paradigma radicaal veranderd. Dat is onder meer toe te schrijven aan een verandering van de behandeling van pulmonale arteriële hypertensie.
In tegenstelling tot pulmonale hypertensie in het algemeen is pulmonale arteriële hypertensie een zeldzame (prevalentie en incidentie respectievelijk minstens 15 gevallen/miljoen en 2,5 gevallen/miljoen/ jaar4), ernstige (mediane overleving zonder behandeling < 3 jaar) en ongeneeslijke aandoening.2 Pulmonale arteriële hypertensie kan idiopathisch of hereditair zijn of kan te wijten zijn aan een toxische stof (bijvoorbeeld amfetaminederivaten of behandeling met dasatinib). Die vormen zijn goed voor ongeveer de helft van de gevallen die in gespecialiseerde centra gezien worden.
In de andere helft van de gevallen is de pulmonale arteriële hypertensie een complicatie van een onderliggende ziekte zoals een bindweefselziekte (vooral systemische sclerose), een aangeboren hartziekte (met al dan niet een eisenmengercomplex), portale hypertensie en hiv-infectie.1 Die ziektes worden echter zelden gecompliceerd met een pulmonale arteriële hypertensie: pulmonale arteriële hypertensie komt voor bij 15 % van de patiënten met een aangeboren hartziekte, bij 1 % van de patiënten met cirrose en bij 0,5 % van de hiv-geïnfecteerde patiënten. Systemische sclerose wordt in 8-10 % van de gevallen gecompliceerd met pulmonale hypertensie. Meestal gaat het om een pulmonale hypertensie door interstitieel longlijden of linkerhartdisfunctie, en slechts in een klein percentage van de gevallen om pulmonale arteriële hypertensie.1, 5
Met andere woorden, pulmonale hypertensie bij een bindweefselziekte is niet altijd een pulmonale arteriële hypertensie. Dat onderscheid is niet onbelangrijk, want in tegenstelling tot de andere vormen van pulmonale hypertensie bestaat er een gevalideerd behandelingsalgoritme voor pulmonale arteriële hypertensie1-3, 6.
1998-2018: 20 jaar evolutie van het therapeutische paradigma bij pulmonale arteriële hypertensie
Evian, 1998 … Tweede wereldsymposium over pulmonale hypertensie
De 80 experts (en een handvol jonge beginnelingen) stellen voor het eerst meerdere therapeutische wegen voor. Toentertijd was er enkel epoprostenol intraveneus, een prostacyclinederivaat, dat voorgeschreven werd bij patiënten met een ernstige idiopathische pulmonale arteriële hypertensie, op grond van twee klinische studies die gunstige hemodynamische en klinische effecten en een daling van de sterfte aangetoond hadden in vergelijking met de standaardbehandeling, in casu aspecifieke behandelingen.1, 6 Maar het opvallendste idee was al om het effect van een multimodale combinatietherapie te onderzoeken.
Venetië, februari 2003 … Derde wereldsymposium over pulmonale hypertensie
Iets meer experts (100), maar vooral (voor de eerste keer) 200 deelnemers. Er werden drie pathofysiologische mechanismen besproken:
- de endothelineweg: een te sterke activering van die weg veroorzaakt een overmatige vasoconstrictie en vasculaire remodellering, die mogelijk tegengegaan worden door endothelinereceptorantagonisten;
- de prostacyclineweg: prostacycline heeft een vaatverwijdend en plaatjesaggregatieremmend effect. Bij een tekort aan prostacycline tanen die effecten. De prostacyclineweg kan gestimuleerd worden door parenterale toediening van prostacyclinederivaten (epoprostenol, iloprost, treprostinil) of orale toediening van prostacyclinereceptoragonisten (selexipag);
- de cGMP-weg: via remming van de afbraak van cyclisch GMP door een type 5-fosfodi-esteraseremmer (PDE5-remmer) of door directe stimulering van guanylaatcyclase (riociguat).1
Rigoureuze, multicentrische, placebogecontroleerde studies hebben die verschillende wegen geëxploreerd gedurende 12-16 weken. Het primaire eindpunt van effectiviteit was de afstand die in 6 minuten kon worden afgelegd. Op grond daarvan werd het eerste orale geneesmiddel in de handel gebracht, meer bepaald bosentan. Dat werd al snel gevolgd door andere geneesmiddelen (sildenafil, treprostinil subcutaan). Het eerste behandelingsalgoritme voor pulmonale arteriële hypertensie werd gepresenteerd tijdens de allereerste editie van de richtlijnen van de Europese Vereniging voor Cardiologie (ESC)7.
De behandeling bestond toen uit toediening van één enkel geneesmiddel gericht tegen een van de bovenvermelde wegen, naargelang van de symptomen volgens de classificatie van de New York Heart Association (NYHA). Een sequentiële combinatietherapie werd enkel aangeraden als de toestand verslechterde (zwakke aanbeveling bij gebrek aan bewijzen).
Daarna verscheen er meer bewijsmateriaal en 5 jaar later werd een nieuw algoritme beschreven in de 2e editie van de richtlijnen, die gezamenlijk door de twee grote Europese wetenschappelijke verenigingen, de ESC en de ERS, opgesteld werden en in 2009 gepubliceerd werden. Als eerstelijnstherapie werd nog altijd een orale monotherapie voorgesteld (of een parenterale monotherapie bij patiënten in NYHA-klasse III/IV), maar de sequentiële combinatietherapie (endothelinereceptorantagonist + PED5-remmer voor de orale combinatietherapie) werd een niveau hoger geklasseerd.8
Tien jaar later werd de 3e editie van de richtlijnen van de ESC/ERS1 voor pulmonale hypertensie gepubliceerd. Op dat ogenblik is de behandeling van pulmonale arteriële hypertensie sterk veranderd en wordt geopteerd voor een totaalaanpak. Het behandelingsparadigma verandert dankzij twee belangrijke aanwinsten:
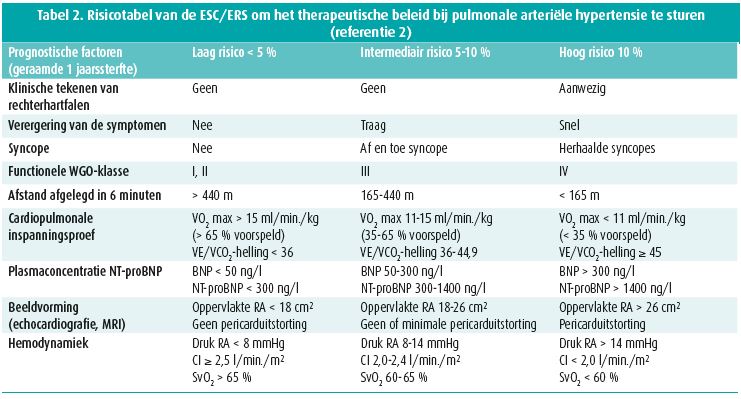
- evaluatie van het risico op grond van klinische criteria, de inspanningstolerantie en de hartfunctie in plaats van louter de NYHA-klasse (tabel 2);
- de sterkste aanbeveling om meteen een combinatietherapie te starten bij
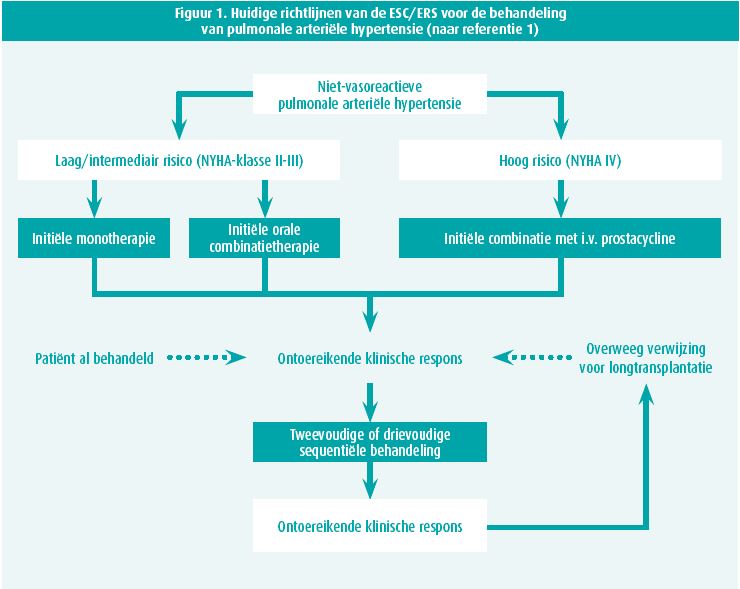
- patiënten met een intermediair risico. Bij evaluatie van het risico op het ogenblik dat de diagnose gesteld is, kunnen de patiënten in drie groepen ingedeeld worden: laag, intermediair en hoog risico (tabel 2). En net zoals bij andere ernstige aandoeningen geldt: hoe hoger het risico, des te krachtiger de behandeling moet zijn. Patiënten die een hoog risico lopen, moeten behandeld worden met een combinatie die onder meer een prostacycline intraveneus bevat.1 Bij minder ernstige vormen (laag of intermediair risico) mag men kiezen tussen een monotherapie of een tweevoudige combinatietherapie per os (figuur 1). Na 4-6 maanden behandeling moet het risico opnieuw geraamd worden met een rechterhartkatheterisatie, om het beleid aan te passen.
Nice, februari 2018 … 6e wereldsymposium over pulmonale hypertensie
Het aantal experts is stabiel (124), maar het aantal deelnemers stijgt en is nu al hoger dan duizend, 1376 om precies te zijn. Moet het behandelingsalgoritme nog gewijzigd worden? Ja, ongetwijfeld … Maar met 42 klinische studies die meerdere behandelingen geëvalueerd hebben bij in totaal meer dan 9 000 patiënten (een record voor een zeldzame ziekte) is dat geen makkelijke taak.9
Een opmerkelijke vaststelling is dat in minder dan 5 jaar meerdere studies met een combinatietherapie uitgetest zijn. Op grond van die resultaten is de behandeling aangepast (tabel 3). Voor de allereerste keer was het eindpunt in die 4 studies niet de afstand die in 6 minuten afgelegd kon worden, maar de incidentie van ernstige accidenten. Het primaire eindpunt verschilde van studie tot studie, maar was altijd een samengesteld eindpunt.
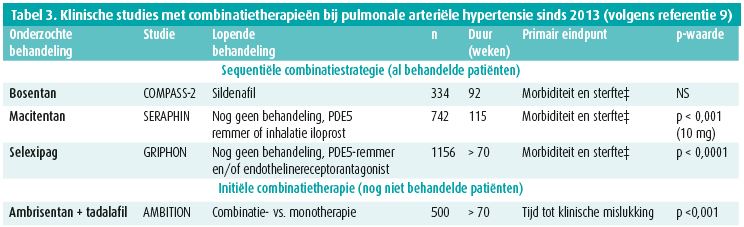
Op grond van de studies SERAPHIN en GRIPHON zijn twee doeltreffende geneesmiddelen in de handel gebracht, namelijk macitentan en selexipag.
De AMBITION-studie is vanuit meerdere oogpunten uniek. Het is de eerste (en tot dusver de enige gepubliceerde) studie die meteen een combinatietherapie met ambrisentan, een endothelinereceptorantagonist, en tadalafil, een PDE5-remmer, heeft vergeleken met ambrisentan of tadalafil in monotherapie10.
Het primaire eindpunt was een samengesteld eindpunt van 'klinisch falen', zijnde overlijden, ziekenhuisopname wegens verergering van de pulmonale arteriële hypertensie, klinische verergering (hogere NYHA-klasse + daling van de in 6 minuten afgelegde afstand met 15 %) en een onvoldoende respons (tekenen van klinische verergering na 6 maanden behandeling). Origineel in deze studie is dat er geen placebogroep was. Voor de eerste keer hebben alle onderzochte groepen een gevalideerde behandeling voor pulmonale arteriële hypertensie gekregen. Het aantal evenementen in de groep die de combinatietherapie kreeg, was 18 %. In de groep die ambrisentan of tadalafil in monotherapie kreeg, was dat 31 %, dus een daling van het relatieve risico met 50 % (hazard ratio 0,5, 95 % betrouwbaarheidsinterval 0,35-0,72, p-waarde < 0,001) en een daling van het absolute risico met 13 %.10 Net zoals in de andere studies1, 9 daalde de mortaliteit niet significant, maar het aantal ziekenhuisopnames wegens verergering van de ziekte wel (daling met 60 %).11
Onafhankelijke voorspellers van een ziekenhuisopname waren:
- behandeling met één enkel geneesmiddel;
- korte loopafstand bij inclusie;
- hogere NT-proBNP-spiegel bij inclusie.11
Bij analyse van het risicoprofiel bleek de NYHA-klasse niet te volstaan om de ernst van de ziekte te ramen. Volgens de REVEAL-score zat slechts 77 % van de patiënten van klasse II in de laagrisicocategorie, tegen 32 % van de patiënten van NYHA-klasse III.12
Naast een evaluatie van het risico op het ogenblik van de diagnose weegt ook het optreden van een klinisch accident tijdens de ziekte-evolutie zwaar op de prognose. Vorsers hebben de gegevensbanken van de studies SERAPHIN (macitentan vs. placebo) en GRIPHON (selexipag vs. placebo) (sequentiële behandeling) uitgebreid geanalyseerd.13
In het eerste geval was het overlijdensrisico bij patiënten die meer dan 3 maanden voor het starten van de behandeling een morbide evenement vertoond hadden, hoger dan bij patiënten zonder morbide evenement (hazard ratio [HR]: 3,39; 95 % betrouwbaarheidsinterval: 1,94-5,92).
In de GRIPHON-studie was het risico ook hoger (HR 4,48; 95 % BI: 2,98-6,73). De observaties na 6 en 12 maanden hebben soortgelijke uitkomsten gegeven, hoewel het risico lager was. Dat wijst erop dat een vroege interventie het risico significanter kan verlagen.13
Combinatietherapie bij pulmonale arteriële hypertensie: hoe eerder, hoe beter
Een jaar na het laatste wereldsymposium zijn er ruimschoots voldoende gegevens om het therapeutische paradigma bij pulmonale arteriële hypertensie opnieuw te wijzigen. Het nieuwe algoritme (figuur 2) benadrukt het belang van starten van een combinatietherapie zodra de diagnose gesteld is, met hetzij een orale combinatie bij patiënten met een laag of intermediair risico, hetzij een combinatie van een orale behandeling en een parenteraal prostacycline. Een monotherapie is enkel nog geïndiceerd in zeldzame situaties, zoals bij bejaarden, patiënten met portopulmonale arteriële hypertensie en patiënten die een zeer laag risico lopen.9
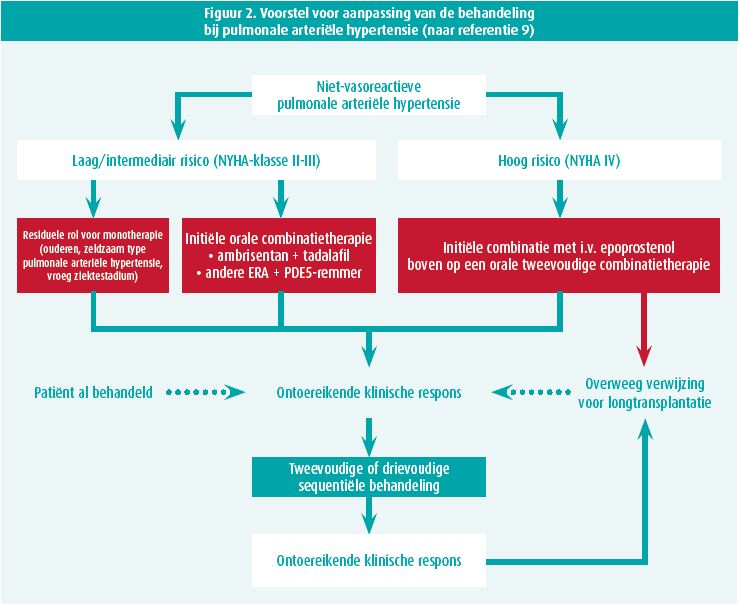
En nu? Hoewel er veel vooruitgang geboekt is, blijft pulmonale arteriële hypertensie een ongeneeslijke ziekte. Daarom lopen er meerdere klinische studies om het therapeutische beleid te verbeteren. Zo is er de TRITON-studie, die een drievoudige combinatietherapie (macitentan + tadalafil + selexipag) vergelijkt met een tweevoudige combinatietherapie (macitentan + tadalafil) bij patiënten bij wie de diagnose recent gesteld is. Het primaire eindpunt is de pulmonale vaatweerstand na 6 maanden behandeling (NCT02558231).
We kijken met spanning uit naar de resultaten van die studie, die allicht in de komende weken wereldkundig gemaakt zullen worden. Ook moeten we nieuwe therapeutische wegen exploreren, zoals een van de 13 werkgroepen op het laatste wereldcongres voorgesteld heeft.14 Met een tiental fase 2- en fase 3-studies wordt verbeten verdergestreden tegen deze ziekte …
Referenties
- Galiè, N., Humbert, M., Vachiéry, J.L., et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J, 2015, 46, 903-975 and Eur Heart J, 2016, 37, 67-119.
- Vachiéry, J.L., Adir, Y., Barberà, J.A., et al. Pulmonary hypertension due to heart diseases. J Am Coll Cardiol, 2013, 62, D100-108.
- Seeger, W., Adir, Y., Barberà, J.A., et al. Pulmonary hypertension due to lung diseases. J Am Coll Cardiol, 2013, 62, D109-116.
- Humbert, M., Sitbon, O., Chaouat, A., et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med, 2006, 173, 1023-1030.
- Vachiéry, J.L., Coghlan, G. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev, 2009, 18, 113, 162-169.
- Vachiéry, J.L., Gaine, S. Challenges in the diagnosis and treatment of pulmonary arterial hypertension. Eur Respir Rev, 2012, 21, 126, 313-320.
- Galiè, N., Torbicki, A., Barst, R.J.B. et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J, 2004, 25, 2243-2278.
- Galiè, N., Hoeper, M.M., Humbert, M., et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J, 2009, 30, 2493-2537.
- Galiè, N., Channick, R., Frantz, B., et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J, 2019, 53 (1), Pii: 1801889. Doi: 10.1183/13993003.01889-2018.
- Galiè, N., Barberà, J.A., Frost, A.E., et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373, 834-844.
- Vachiéry, J.L., Galiè, N., Barberá, J.A., et al. Initial combination therapy with ambrisentan+ tadalafil on pulmonary arterial hypertension- related hospitalization in the AMBITION trial. J Heart Lung Transplant, 2019, 38, 194-202.
- Frost, A.E., Hoper, M.M., Barbera, J.A., et al. Risk-stratified outcomes with initial combination therapy in pulmonary arterial hypertension: Application of the REVEAL risk score. J Heart Lung Transplant, 2018, 37, 1410-1417.
- McLaughlin, V.V., Hoeper, M.M., Channick, R.N., et al. Pulmonary Arterial Hypertension- Related Morbidity Is Prognostic for Mortality. J Am Coll Cardiol, 2018, 71, 752-763.
- Sitbon, O., Gomberg-Maitland, M., Granton, J., et al. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J, 2019, Jan 24;53. pii: 1801908. doi: 10.1183/13993003.01908-2018.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.