ESC-congresverslag
Op dit symposium werden de nieuwe richtlijnen van de Europese Vereniging voor Cardiologie1 besproken, met in het bijzonder: de nieuwe definitie van pulmonale hypertensie; de diagnostiek en evaluatie van het risicoprofiel van patiënten om hen beter te behandelen en de prognose op lange termijn te verbeteren; het belang van de verschillende therapeutische mogelijkheden en van een tweevoudige combinatietherapie.
De diagnose van pulmonale hypertensie (PH) stoelt op een hemodynamische definitie: een gemiddelde druk in de longslagader (PAPm) bij rechterhartkatheterisatie ≥ 20 mmHg in rust. Door de pulmonale wigdruk te meten kan een precapillaire PH (wigdruk ≤ 15 mmHg) worden onderscheiden van een postcapillaire PH (> 15 mmHg). We spreken van precapillaire pulmonale hypertensie bij een patiënt met een PAPm > 20 mmHg, een wigdruk < 15 mmHg en een weerstand van de longslagaders > 2 Wood-eenheden. Als de wigdruk > 15 mmHg is, onderscheiden we twee vormen:
- geïsoleerde postcapillaire pulmonale hypertensie of IpcPH (PAPm > 20 mmHg, wigdruk > 15 mmHg en weerstand van de longslagaders ≤ 2 Wood-eenheden);
- gecombineerde pre- en postcapillaire PH of CpcPH (PAPm > 20 mmHg, wigdruk > 15 mmHg en weerstand van de longslagaders > 2 Wood-eenheden).
Die lagere afbreekwaarde “heeft echter nog niet geleid tot nieuwe therapeutische richtlijnen”, stelt de richtlijn, omdat de werkzaamheid van de behandeling van pulmonale arteriële hypertensie bij patiënten met een gemiddelde druk in de longslagader (PAPm) van 21-24 mmHg niet bekend is. Het is dus belangrijk eraan te herinneren dat een behandeling enkel wordt aanbevolen als de werkzaamheid ervan bewezen is. Dat betekent een PAPm > 25 mmHg.
Pulmonale arteriële hypertensie (groep 1) wordt gedefinieerd als een combinatie van hemodynamische criteria (PAPm > 20 mmHg, pulmonale capillaire bloeddruk ≤ 15 mmHg en weerstand van de longslagaders > 2 WE) en klinische criteria (geen klinische aandoening die compatibel is met een andere groep van precapillaire pulmonale hypertensie: uitsluiten van andere oorzaken zoals een longziekte [groep 3], een trombo-embolie [groep 4] of een andere ziekte [groep 5]).2
Het is belangrijk om te onthouden dat PAH (groep 1) een zeldzame oorzaak van pulmonale hypertensie is in een niet-geselecteerde populatie (15-60/1,106 volwassenen). Bijna driekwart van de gevallen van pulmonale hypertensie wordt veroorzaakt door linkerhartfalen (groep 2). De op een na belangrijkste oorzaak van pulmonale hypertensie zijn longziekten die een gebrekkige oxygenatie van het bloed veroorzaken (groep 3).3 Er zijn meerdere oorzaken van pulmonale arteriële hypertensie (PAH): idiopathische, familiale, als gevolg van inname van geneesmiddelen/toxische stoffen en PAH in samenhang met een bindweefselziekte (systemische sclerose), hiv-infectie, portale hypertensie en een aangeboren hartziekte. De nieuwe richtlijnen onderscheiden nu ook een groep oudere patiënten met pulmonale arteriële hypertensie én comorbiditeit.
Op het symposium hebben de experts ook de sleutelrol van een echocardiografie besproken en de verschillende te onderzoeken parameters doorgenomen. Ze hebben daarbij gepleit voor de bepaling van een nieuwe parameter, die de koppeling tussen het rechterventrikel en de longslagader weerspiegelt: de verhouding tussen de TAPSE en de geraamde systolische druk in de longslagader.4 Met een echocardiografie kunnen de patiënten in drie groepen worden ingedeeld: lage, intermediaire of hoge waarschijnlijkheid van pulmonale hypertensie en op grond daarvan kan de patiënt worden verwezen voor aanvullende onderzoeken (tabel 1, figuur 1).
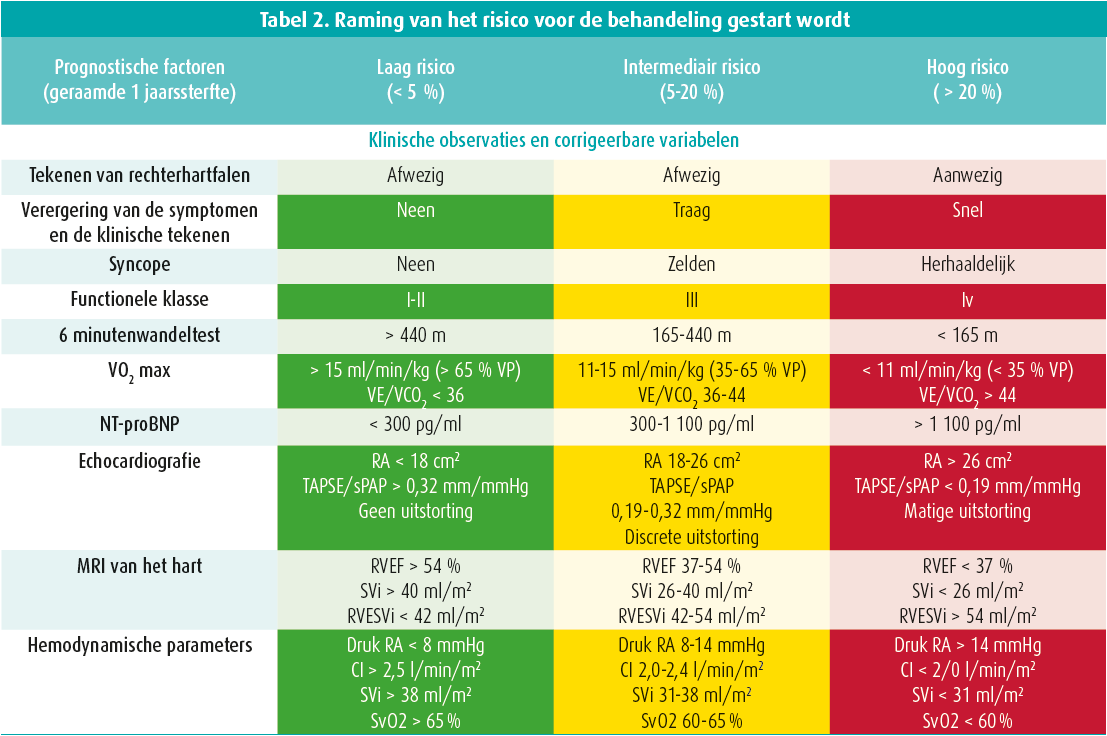
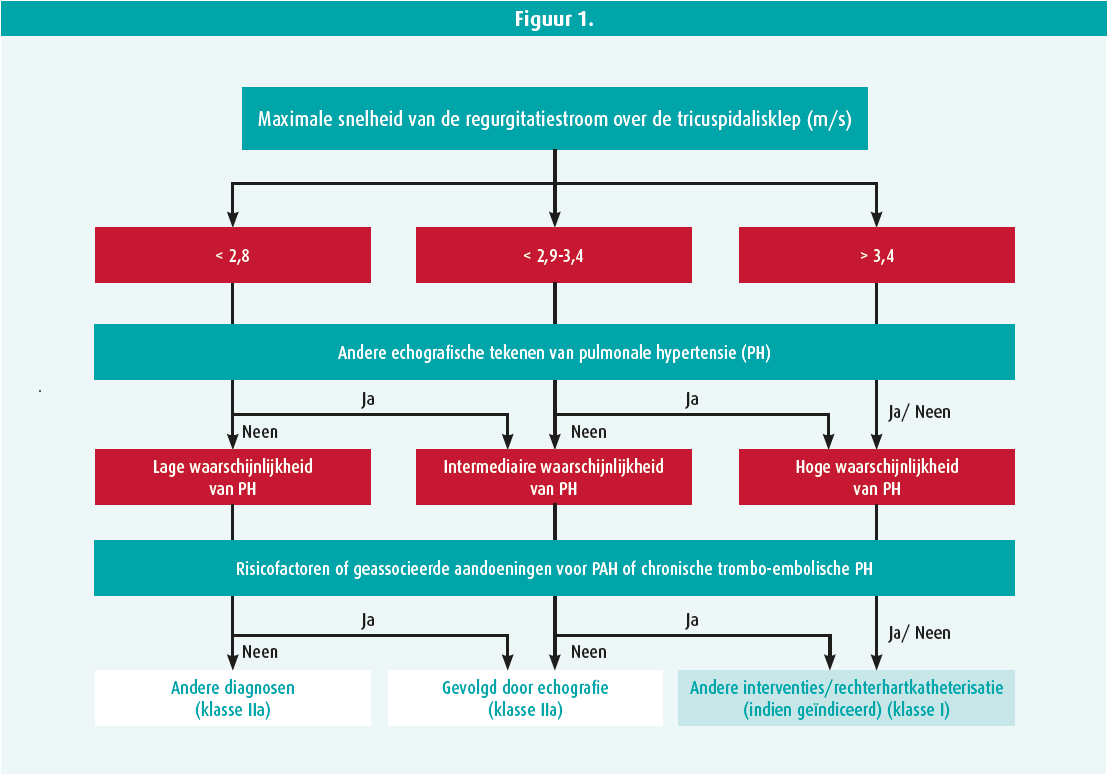
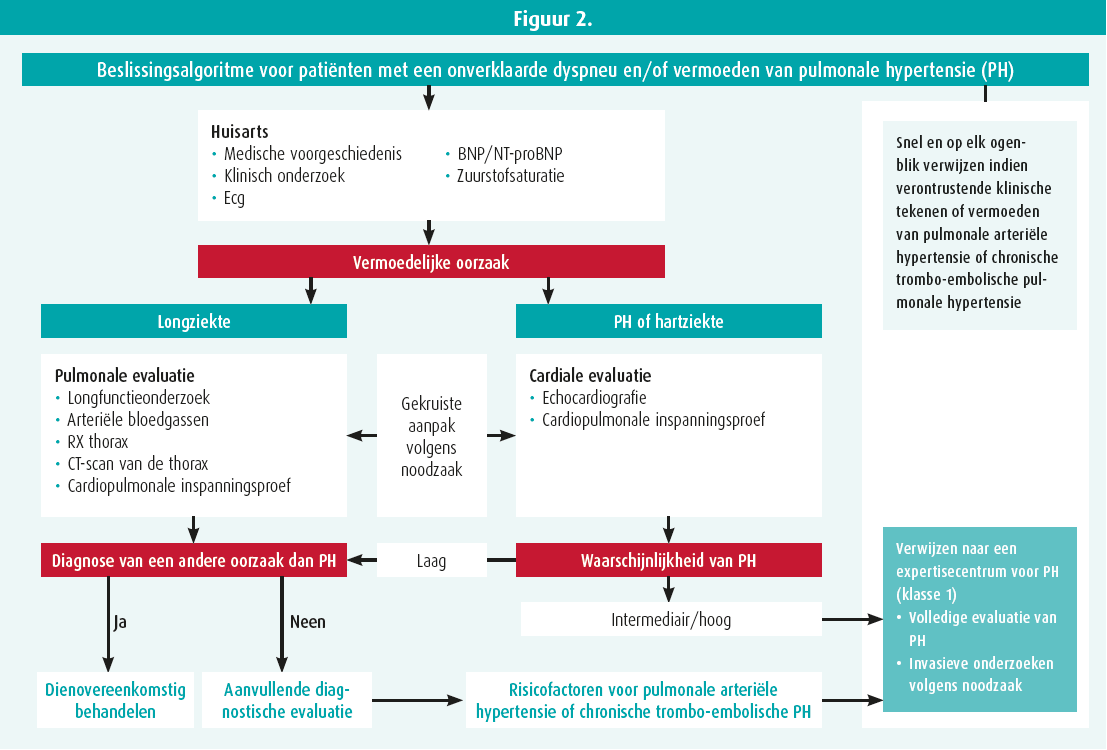
Daarna hebben ze het diagnostische algoritme (figuur 2) toegelicht. Zodra een diagnose van pulmonale arteriële hypertensie gesteld is, moet het risico van de patiënt worden geëvalueerd (tabel 2, vroege stratificatie van het risico in drie groepen) en op grond daarvan wordt de behandeling bepaald. Met die tabel kan het overlijdensrisico na een jaar geraamd worden (tot 20 % in geval van een hoog risico).5
Er bestaan nu geneesmiddelen die gericht zijn tegen de grote drie wegen die meespelen bij de pathofysiologie van pulmonale arteriële hypertensie (de endothelineweg, de NO-weg en de prostacyclineweg).6 Het therapeutische beleid wordt gebaseerd op de ernst van de ziekte en het initiële risico van de patiënt (figuur 3). In de gerandomiseerde, gecontroleerde studies AMBITION7 en SERAPHIN8 werd aangetoond dat een combinatietherapie met een endothelinereceptorantagonist en een fosfodiesteraseremmer de symptomen en de prognose van de patiënten verbetert in vergelijking met een monotherapie.
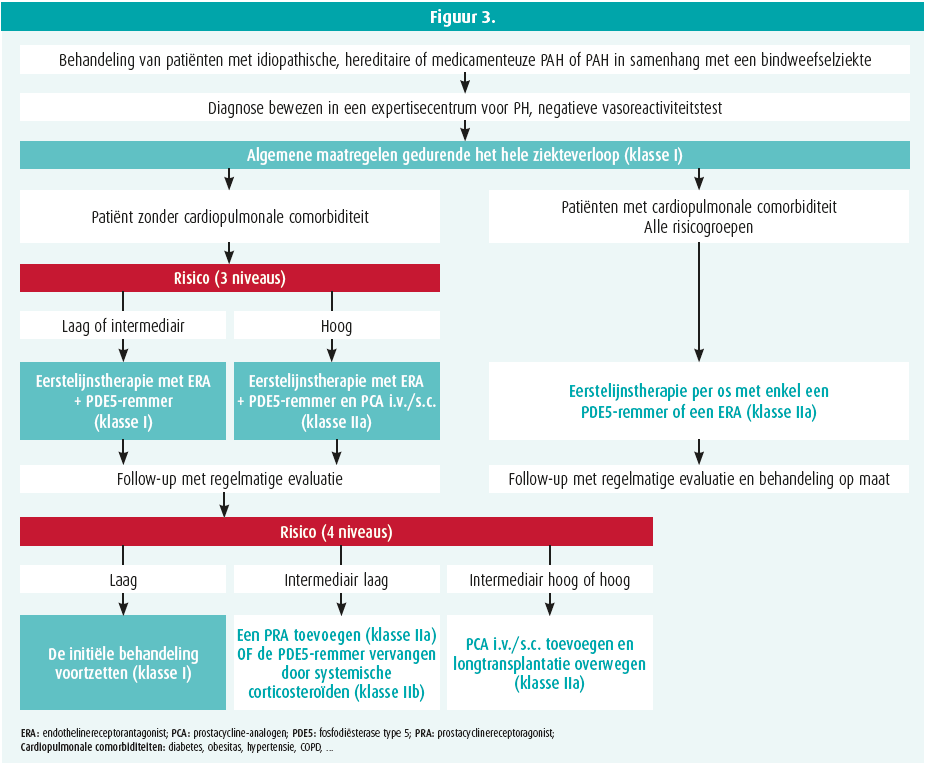
De experts hebben ook benadrukt dat de patiënt al na drie maanden opnieuw moet worden geëvalueerd en dat het risico dan moet worden geraamd, ditmaal aan de hand van een risicotabel met vier strata (tabel 3).9 Als de patiënt niet verbetert tot het niveau van de laagrisicogroep, moet de behandeling worden geïntensiveerd. De prognose is immers minder goed als de patiënten in de intermediair-risicogroep blijven zitten. De behandeling moet dan worden opgedreven.
- Laag intermediair risico: de toevoeging van een orale prostacyclineagonist moet worden overwogen (GRIPHON-studie, 37 % lager relatief risico op ziekteprogressie met een drievoudige dan met een tweevoudige combinatietherapie)10 (klasse IIA) of de vervanging van de fosfodi-esteraseremmer door een geneesmiddel dat het guanylaatcyclase stimuleert kan ook worden overwogen (REPLACE-studie) (klasse IIB);11
- Intermediair hoog of hoog risico: prostacycline i.v. of s.c. moet worden overwogen, evenals de patiënt verwijzen voor longtransplantatie (klasse IIA).
De behandeling van pulmonale arteriële hypertensie is een grote klinische uitdaging. Er moet snel een juiste diagnose worden gesteld en er moet tijdig een specifieke behandeling worden gestart. Vaak wordt de diagnose pas in een gevorderd en vaak irreversibel stadium gesteld. De prognose is dan uiterst slecht.
Referenties
- Humbert, M., Kovacs, G., Hoeper, M.M., Badagliacca, R., Berger, R.M.F., Brida, M. et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J, 2022, 30, 2200879.
- Rosenkranz, S., Gibbs, J.S., Wachter, R., De Marco, T., Vonk-Noordegraaf, A., Vachiéry, J.L. Left ventricular heart failure and pulmonary hypertension. Eur Heart J, 2016, 37 (12), 942-945.
- Gabbay, E., Yeow, W., Playford, D., Pulmonary arterial hypertension (PAH) is an uncommon cause of pulmonary Hypertension (PH) in an unselected population: the Armadale echocardiography study. Am J Resp Crit Care Med, 2007, 175, A713.
- Tello, K., Axmann, J., Ghofrani, H.A., Naeije, R., Narcin, N., Rieth, A. et al. Relevance of the TAPSE/PASP ratio in pulmonary arterial hypertension. Int J Cardiol, 2018, 266, 229-223.
- Hoeper, M.M., Kramer, T., Pan, Z., Eichstaedt, C.A., Spiesshoefer, J., Benjamin, N. et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J, 2017, 50 (2), 1700740.
- Lau, E.M.T., Giannoulatou, E., Celermajer, D.S., Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol, 2017, 14 (10), 603-614.
- Galiè, N., Barberà, J.A., Frost, A.E., Ghofrani, H.A., Hoeper, M.M., McLaughlin, V.V. et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373 (9), 834-844.
- Pulido, T., Adzerikho, I., Channick, R.N., Delcroix, M., Galiè, N., Ghofrani, H.A. et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med, 2013, 369 (9), 809-818.
- Boucly, A., Weatherald, J., Savale, L., de Groote, P., Cottin, V., Prévot, G. et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J, 2022, 59 (6), 2102419.
- Coghlan, J.G., Channick, R., Chin, K., Di Scala, L., Galiè, N., Ghofrani, H.A. et al. Targeting the Prostacyclin Pathway with Selexipag in Patients with Pulmonary Arterial Hypertension Receiving Double Combination Therapy: Insights from the Randomized Controlled GRIPHON Study. Am J Cardiovasc Drugs, 2018, 18 (1), 37-47.
- Hoeper, M.M., Al-Hiti, H., Benza, R.L., Chang, S.A., Corris, P.A., Gibbs, J.S.R. et al. Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open-label, randomised controlled trial. Lancet Respir Med, 2021, 9 (6), 573-584.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.