Compte rendu du congrès de l'ESC
Les objectifs de ce symposium étaient de parcourir les nouvelles recommandations de la Société européenne de cardiologie1, en particulier : revoir la nouvelle définition de l'hypertension pulmonaire ; les outils à utiliser pour faire le diagnostic et comment évaluer le profil de risque du patient pour mieux le traiter et améliorer son pronostic à long terme ; reconnaître l'importance des différentes possibilités thérapeutiques et la place de la bithérapie.
Le diagnostic d'hypertension pulmonaire repose sur une définition hémodynamique avec une pression artérielle pulmonaire moyenne (PAPm) mesurée au cathétérisme cardiaque droit ≥ 20 mmHg au repos. La mesure de la pression artérielle pulmonaire occluse (PAP0) permet la distinction entre une origine pré-capillaire (PAPO ≤ 15 mmHg) ou post-capillaire (> 15 mmHg). On parlera d'hypertension pulmonaire précapillaire en présence d'une PAPm > 20 mmHg, avec une PAPO < 15 mmHg et des résistances vasculaires pulmonaires (RVP) > 2 unités Wood. Si la PAPO est > 15 mmHg, 2 formes sont identifiées :
- L'hypertension pulmonaire postcapillaire isolée ou IpcPH (PAPm > 20 mmHg, PAPO > 15 mmHg et RVP ≤ 2 unités Wood) ;
- L'hypertension pulmonaire combinée, pré- et post-capillaire ou CpcPH (PAPm > 20 mmHg, PAPO > 15 mmHg et RVP > 2 unités Wood).
Cependant, ce seuil inférieur « ne se traduit pas encore par de nouvelles recommandations thérapeutiques » , indique la directive, car l'efficacité du traitement de l'hypertension artérielle pulmonaire chez les patients dont la pression artérielle pulmonaire moyenne (PAPm) est de 21-24 mmHg est inconnue. Il est donc important de rappeler qu'actuellement le traitement n'est recommandé que lorsque nous avons la preuve de son efficacité, ce qui signifie une PAPm > 25 mmHg.
L'hypertension artérielle pulmonaire (groupe 1) se définit donc par l'association de critères hémodynamiques (PAPm > 20 mmHg, PAPO ≤ 5 mmHg et résistances vasculaires pulmonaires > 2 UW) et cliniques (absence de conditions cliniques compatibles avec un autre groupe d'hypertension pulmonaire pré-capillaire : exclusion de toute autre cause comme une maladie pulmonaire [groupe 3], une maladie thrombo-embolique [groupe 4] ou une autre maladie [groupe 5]).2
Il est important de rappeler que l'HTAP (groupe 1) est une cause rare d'HTP dans une population non sélectionnée prévalence (15-60/1,106 adultes). En effet, l'insuffisance cardiaque gauche (groupe 2) est la cause de près des trois-quarts des hypertensions pulmonaires. Les maladies pulmonaires responsables d'un défaut d'oxygénation du sang (groupe 3) sont la seconde cause des hypertensions pulmonaires.3 L'hypertension artérielle pulmonaire (HTAP) rassemble plusieurs formes, principalement : les formes idiopathiques, familiales, secondaires à la prise de médicaments/toxiques et les formes associées : aux connectivites (sclérose systémique), au virus du VIH, à l'hyper tension portale ou aux cardiopathies congénitales. Les nouvelles recommandations identifient également aussi maintenant un groupe de patients plus âgés, présentant une HTAP avec comorbidités.
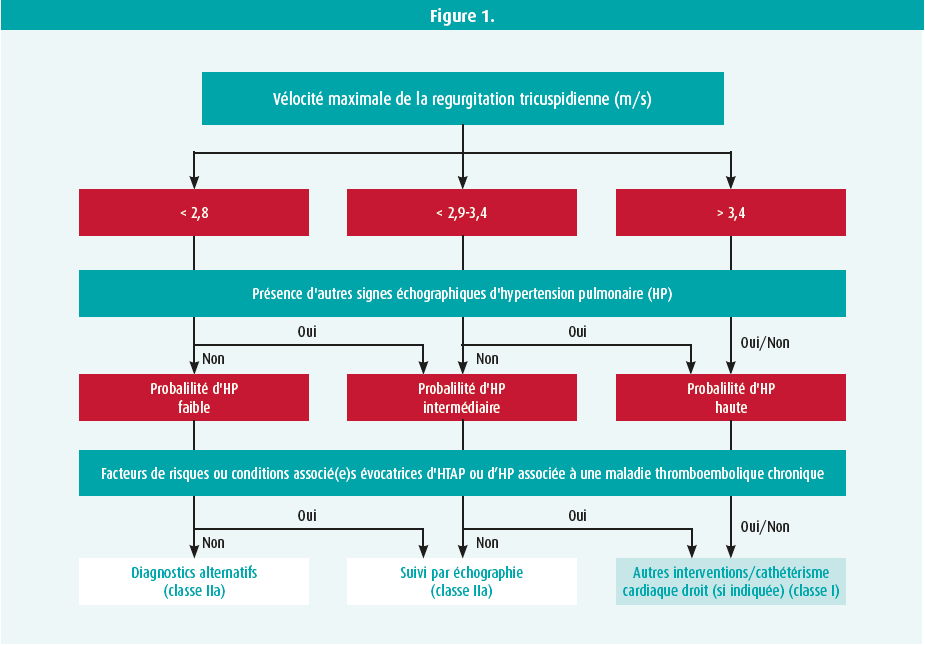
Les experts réunis pendant ce symposium ont également insisté sur le rôle clé de l'échocardiographie et ont revus les différents paramètres à évaluer, en insistant sur un nouveau paramètre reflétant le couplage ventricule droit/artère pulmonaire : le rapport entre le TAPSE et l'estimation de la pression pulmonaire systolique.4 L'échocardiographie nous permettra donc de classer les patients en 3 groupes : probabilité d'hypertension pulmonaire faible, intermédiaire ou élevée, et donc de référer le patient pour bilan complémentaire (tableau 1, figure 1).

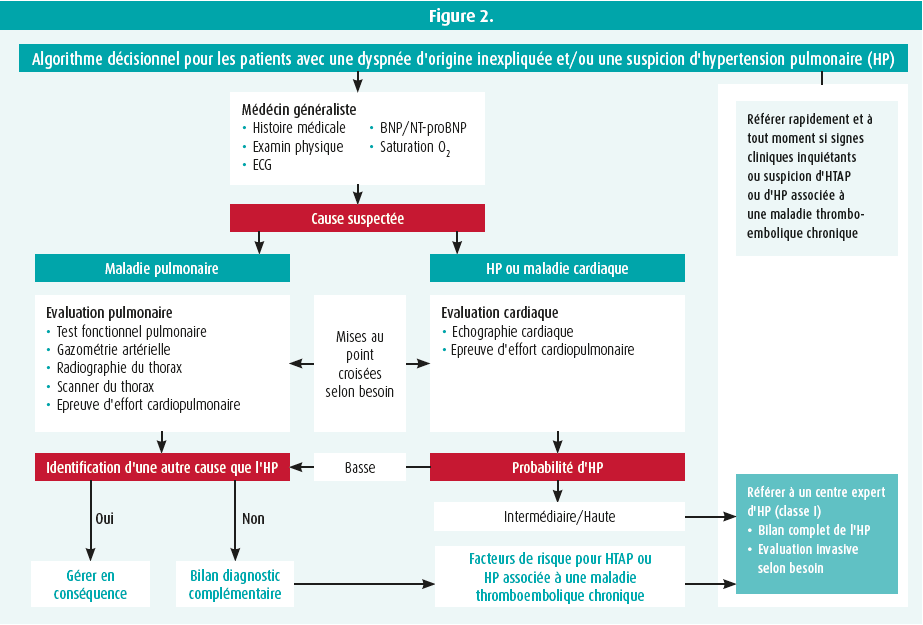
Ils ont ensuite revu l'algorithme diagnostic (figure 2). Une fois le diagnostic d'hyper tension artérielle pulmonaire posé, il sera important d'évaluer le risque du patient (tableau 2, stratification précoce du risque à 3 strates) afin de proposer la thérapeutique la plus adaptée. Cette table permet d'estimer le risque de mortalité à 1 an (jusque 20 % de mortalité à 1 an pour les patients dans la catégorie à haut risque).5
Des traitements sont actuellement disponibles et cibles les 3 grandes voies impliquées dans la physiopathologie de l'HTAP (la voie de l'endothéline, la voie du NO et la voie des prostacyclines).6 La stratégie thérapeutique est basée sur la sévérité de la maladie et le risque initial du patient (figure 3). Les études cliniques randomisées contrôlées AMBITION7 et SERAPHIN8 ont en effet démontré qu'une thérapie combinée par antagonistes des récepteurs de l'endothéline et inhibiteurs des phosphodiestérases améliorent les symptômes et le pronostic des patients en comparaison à une monothérapie.
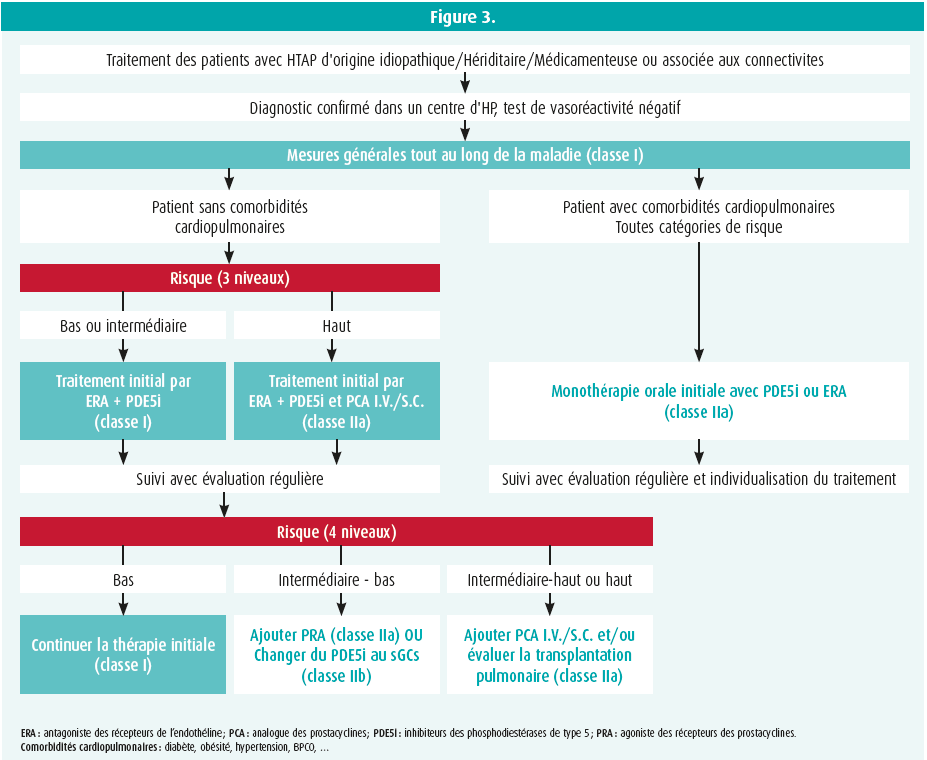
Enfin, les experts ont insisté sur l'importance d'une réévaluation précoce du patient (à 3 mois) et de son profil de risque en utilisant cette fois une table de risque avec 4 strates (tableau 3).9 Le traitement doit être intensifié si le patient n'atteint pas la catégorie de risque bas. En effet, les patients restant dans une catégorie de risque intermédiaire ou haut ont un moins bon pronostic et nécessitent donc un traitement renforcé.
- Profil intermédiaire bas : l'ajout d'un agoniste des prostacyclines orales devrait être envisagé (étude GRIPHON, réduction de 37 % du risque relatif de progression de la maladie sous triple thérapie par rapport à une bithérapie)10 (classe IIA) ou le remplacement des inhibiteurs des phosphodiestérases par un stimulateur de la guanylate cyclase peut également être envisagé (étude REPLACE) (classe IIB) ;
- Profil intermédiaire haut ou haut : l'instauration de prostacyclines par voie IV ou SC doit être envisagé, de même référer le patient pour évaluation pour la transplantation pulmonaire (classe IIA).

Le management de l'HTAP représente un défi clinique majeur à la fois pour poser un diagnostic précis et précoce mais aussi pour instaurer un traitement spécifique à temps. Trop souvent tardif, le diagnostic à un stade avancé et souvent irréversible s'associe à un pronostic extrêmement sévère.
Références
- Humbert, M., Kovacs, G., Hoeper, M.M., Badagliacca, R., Berger, R.M.F., Brida, M. et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J, 2022, 30, 2200879.
- Rosenkranz, S., Gibbs, J.S., Wachter, R., De Marco, T., Vonk-Noordegraaf, A., Vachiéry, J.L. Left ventricular heart failure and pulmonary hypertension. Eur Heart J, 2016, 37 (12), 942-945.
- Gabbay, E., Yeow, W., Playford, D., Pulmonary arterial hypertension (PAH) is an uncommon cause of pulmonary Hypertension (PH) in an unselected population: the Armadale echocardiography study. Am J Resp Crit Care Med, 2007, 175, A713.
- Tello, K., Axmann, J., Ghofrani, H.A., Naeije, R., Narcin, N., Rieth, A. et al. Relevance of the TAPSE/PASP ratio in pulmonary arterial hypertension. Int J Cardiol, 2018, 266, 229-223.
- Hoeper, M.M., Kramer, T., Pan, Z., Eichstaedt, C.A., Spiesshoefer, J., Benjamin, N. et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J, 2017, 50 (2), 1700740.
- Lau, E.M.T., Giannoulatou, E., Celermajer, D.S., Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol, 2017, 14 (10), 603-614.
- Galiè, N., Barberà, J.A., Frost, A.E., Ghofrani, H.A., Hoeper, M.M., McLaughlin, V.V. et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373 (9), 834-844.
- Pulido, T., Adzerikho, I., Channick, R.N., Delcroix, M., Galiè, N., Ghofrani, H.A. et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med, 2013, 369 (9), 809-818.
- Boucly, A., Weatherald, J., Savale, L., de Groote, P., Cottin, V., Prévot, G. et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J, 2022, 59 (6), 2102419.
- Coghlan, J.G., Channick, R., Chin, K., Di Scala, L., Galiè, N., Ghofrani, H.A. et al. Targeting the Prostacyclin Pathway with Selexipag in Patients with Pulmonary Arterial Hypertension Receiving Double Combination Therapy: Insights from the Randomized Controlled GRIPHON Study. Am J Cardiovasc Drugs, 2018, 18 (1), 37-47.
- Hoeper, M.M., Al-Hiti, H., Benza, R.L., Chang, S.A., Corris, P.A., Gibbs, J.S.R. et al. Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open-label, randomised controlled trial. Lancet Respir Med, 2021, 9 (6), 573-584.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.