De quels patients s'agit-il?
L'hypertension pulmonaire est définie comme une pression moyenne au repos dans l'artère pulmonaire égale ou supérieure à 25 mmHg. Cette pathologie peut être provoquée par des problèmes cardiaques gauches (valvulopathies, HFrEF/ pEF, obstruction de la chambre de chasse …), des affections pulmonaires/ une hypoxémie (BPCO, atteinte interstitielle, SAOS …), des embolies pulmonaires chroniques ainsi que toute une série d'affections plus rares.
Si la PCW est basse (15 mmHg ou moins) et la résistance vasculaire pulmonaire élevée (plus de 3 unités Wood), on parle d'hypertension pulmonaire précapillaire. Ensuite, au moyen d'examens techniques, nous devons exclure des cardiopathies gauches à l'origine de l'hypertension, des affections pulmonaires, des embolies pulmonaires chroniques et une série d'autres affections. S'il s'avère qu'un patient souffrant d'hypertension pulmonaire précapillaire ne présente aucune des affections susmentionnées, il souffre alors d'hypertension artérielle pulmonaire (HAP). Au sein de ce groupe d'HAP, il arrive régulièrement que nous ne trouvions pas de cause univoque à l'affection (forme idiopathique). Néanmoins, l'HAP peut être due à des affections rhumatismales (essentiellement la sclérodermie), hépatiques (surtout en cas d'hypertension portale), infectieuses (VIH et schistosomiase) et cardiaques congénitales (surtout les anomalies du septum/shunts). Enfin, des formes génétiques et toxiques sont également bien connues. Les affections qui répondent à la définition de l'HAP sont caractérisées par des anomalies semblables au niveau de la microcirculation pulmonaire, qui constituent la cible pour le traitement médicamenteux (ESC/ERS guidelines on pulmonary hypertension. Galiè, EHJ, 2015, 37, 67-119).
Quel est le pronostic de l'HAP et comment évaluons-nous le risque?
Sans traitement, environ 70 % des patients décèdent dans les 3 ans. La survie dépend partiellement de la cause sous-jacente de l'HAP: l'HAP liée à la sclérodermie est associée au pronostic le plus sombre, et l'HAP due à une cardiopathie congénitale, considérée à partir du moment du diagnostic, est en moyenne associée à une survie plus longue.
L'estimation individuelle du risque de mortalité à 1 an se pratique sur la base d'une évaluation multiparamétrique, constituée des éléments suivants:
- symptômes: classe de l'OMS, évolution et syncopes éventuelles;
- examen clinique: signes de défaillance cardiaque droite;
- capacité à l'effort: test de marche de 6 minutes ou VO2 max et pente VE/ VCO2 à l'ergospirométrie;
- (NT-pro-)BNP;
- paramètres échocardiographiques: surface auriculaire droite, épanchement péricardique;
- paramètres hémodynamiques invasifs: pression OD, index cardiaque et saturation veineuse mixte.
En fonction du résultat, on attribue un score de risque faible, intermédiaire ou élevé pour chaque paramètre. La combinaison de tous ces paramètres donne une idée du risque lors de l'évaluation initiale, comme l'illustre la figure 1.
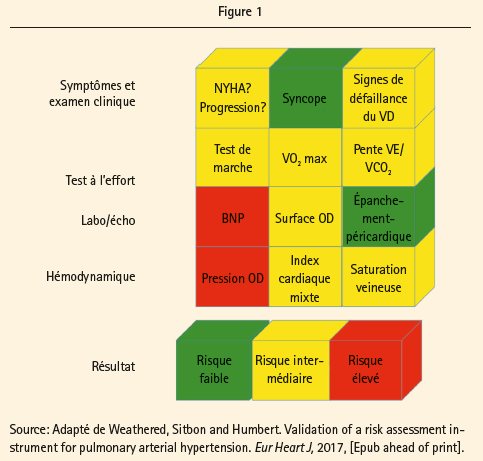
Chez les patients inclus dans le groupe à risque faible, le risque de décès est inférieur à 5 %, tandis qu'il est respectivement égal à 5-10 % et >10 % pour les groupes à risque intermédiaire et élevé. L'importance de l'estimation du risque, tant au départ que lors du suivi sous traitement, a été commentée en détail par le Prof. Humbert. La figure 2 illustre l'évolution éventuellement favorable sous traitement, mais aussi le risque de progression de la pathologie sous-jacente, les comorbidités, l'âge croissant et la complexité du traitement. Les recommandations de l'ESC préconisent une évaluation de l'état clinique avec test de marche tous les 3-6 mois, et une estimation annuelle complète du risque, et ce, même chez les patients stables. Lors du vote du public, il est apparu que 10 % des médecins présents ne réalisent pas de suivi systématique, et que 20 % ne pratiquent des tests qu'en cas de détérioration clinique, ce qui est passablement discordant par rapport à ce que préconisent les recommandations.
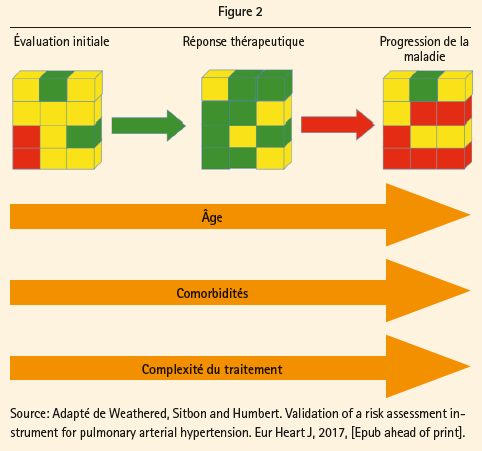
Le but du traitement est non seulement d'améliorer les symptômes et la capacité à l'effort, ce qui constitue souvent le critère d'évaluation des études cliniques, mais en outre de maintenir ou d'atteindre un statut de risque faible tel qu'il peut être estimé sur la base des paramètres cités plus haut. Le statut de risque faible est en effet associé à une meilleure survie et à un risque moindre d'hospitalisation. L'obtention d'un risque intermédiaire est insuffisante, poursuit encore le Prof. Humbert. Ce propos est étayé à l'aide de données très récentes issues de vastes registres français, suédois et allemands (Boucly, A., ERJ, 2017; Kylhammar, EHJ, 2017; Hoeper, ERJ, 2017). La survie dans la catégorie de risque faible est supérieure à 80 % à 5 ans, mais elle diminue à 60 % dans le groupe à risque intermédiaire, tandis que le groupe à risque élevé ne connaît que 10-40 % de survie sans transplantation à 5 ans. Malheureusement, en réalité, seuls 23 % de la population souffrant d'HAP se trouvent dans la catégorie de risque faible au moment du diagnostic, et ce chiffre atteint 29 % au cours du suivi sous traitement, a encore précisé le Prof. Rosenkranz. La plupart des patients souffrant d'HAP courent donc toujours un risque intermédiaire à élevé, et ce, en dépit d'un traitement.
Quelles sont les options médicamenteuses disponibles?
Pour le moment, nous disposons de médicaments qui agissent sur une des 3 voies suivantes:
- la voie du NO: inhibiteurs de la phosphodiestérase de type 5 (PD5i) ou riociguat, un agent stimulant la guanylate cyclase;
- la voie de l'endothéline: antagonistes des récepteurs de l'endothéline (ERA);
- la voie de la prostacycline: prostanoïdes ou sélexipag, un agoniste des récepteurs IP.
Tant au début de la prise en charge que lors de la mise en oeuvre du traitement, les décisions thérapeutiques sont individualisées sur la base de la catégorie de risque, de l'évolution, de l'affection sousjacente, des comorbidités éventuelles et de l'âge du patient.
Pourquoi un traitement combiné?
Étant donné que seule une minorité des patients souffrant d'HAP se trouvent dans le groupe à faible risque au bout d'un an, en dépit du traitement instauré, il semble judicieux de combiner des médicaments qui agissent sur des voies différentes, afin d'obtenir un meilleur résultat. L'étude SERAPHIN a indiqué que l'ajout de macitentan, un ERA, chez les patients déjà traités au moyen d'un PD5i, entraîne une réduction significative du risque du critère d'évaluation combiné, consistant en moment du décès, septostomie, transplantation pulmonaire, instauration d'un traitement par prostanoïdes ou aggravation de l'HAP. Dans l'étude AMBITION, on a également constaté une réduction du risque si on débutait d'emblée par un traitement combiné, consistant en tadalafil (un PD5i) et ambrisentan (un ERA), comparativement à une monothérapie par un de ces produits. Bien que le résultat du traitement combiné se révèle meilleur que celui de la monothérapie, plus de 20 % des patients du groupe combiné ont présenté un événement dans un délai de 2 ans, et ce, tant dans l'étude SERAPHIN que dans AMBITION (Seraphin: Pulido, NEJM, 2013, 369, 809-818. Ambition: Galiè, NEJM, 2015, 373, 834-844).
Triple thérapie: pour qui, quand, et quels composants?
Jusqu'à présent, l'utilisation d'un triple traitement combiné était relativement rare, a démontré le Prof. Rosenkranz: seuls 4 % des patients souffrant d'HAP en Europe ont bénéficié d'une triple thérapie ces dernières années, contre 7 % aux États-Unis et 17 % au Japon. En dépit du fait que les recommandations préconisent un traitement au moyen d'un prostanoïde parentéral dans la classe à haut risque, plus de 33 % des patients à haut risque, souffrant d'HAP, ne reçoivent pas ce traitement. Lors du vote demandant si la triple thérapie fait partie de l'arsenal thérapeutique de routine des médecins présents, il est apparu que seule la moitié d'entre eux dispose de cette possibilité, un quart d'entre eux n'a pas accès aux prostanoïdes ou au sélexipag, et un quart y a peut-être accès, mais n'utilise pas de triple thérapie en pratique.
Dans le sous-groupe de l'étude GRIPHON, lors de laquelle on a ajouté du sélexipag, un agoniste des récepteurs IP, en plus d'un traitement combiné déjà en cours par un ERA et un PD5i, on a de nouveau observé une réduction significative du risque, exprimée en termes de décès ou d'une complication liée à l'HAP. En outre, dans l'étude GRIPHON, on a constaté que la réduction du risque est aussi importante, que le sélexipag soit administré en monothérapie, ajouté à un ERA ou un PD5i, ou en plus de la combinaison d'un ERA et d'un PD5i (Griphon Sitbon, NEJM, 2015, 373, 2522-2533). Ceci fournit un argument supplémentaire en faveur de l'inhibition concomitante de plusieurs voies en cas d'HAP.
Le public a réagi de manière hétérogène lorsqu'on lui a demandé s'il donnerait une triple thérapie aux patients souffrant d'HAP, dans le groupe à risque intermédiaire: 25 % instaureraient d'emblée une triple thérapie (alors qu'on ne dispose pas encore de données en faveur d'un traitement upfront), 25 % n'envisageraient pas de triple thérapie et 50 % donneraient séquentiellement une triple thérapie, si nécessaire. L'étude TRITON examine actuellement si l'instauration immédiate d'une triple thérapie entraîne une amélioration hémodynamique ou physique, comparativement à une double thérapie, chez des patients à risque faible à intermédiaire.
Enfin, on a encore rappelé qu'en cas de réponse inadéquate à un traitement combiné, les patients ad hoc doivent être réorientés en vue d'une transplantation pulmonaire. La survie médiane après une transplantation pulmonaire pour une HAP idiopathique atteint en effet 6 petites années, ce qui semble un peu mieux que le traitement purement médicamenteux dans le groupe à haut risque.
En résumé, cette session a tout d'abord illustré l'importance exceptionnelle d'un suivi multiparamétrique systématique minutieux en cas d'HAP, et la triple thérapie semble judicieuse dans les cas suivants:
- d'emblée pour un patient à haut risque, souffrant d'HAP (prostanoïde IV);
- séquentiellement pour un patient souffrant d'HAP, à risque intermédiaire, qui n'atteint pas le statut de faible risque sous traitement par un PD5i/du riociguat et un ERA (sélexipag PO);
- séquentiellement en cas d'aggravation de l'HAP (ajout de/passage à un prostanoïde IV).
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.