Symposium satellite - orateurs: Pr Fabrice Bauer, Pr Valerie Mc Laughlin, Pr Jean-Luc Vachiéry et Pr Adam Torbicki
Introduction
Ce symposium satellite s'est ciblé sur la prise en charge optimale de l'hypertension artérielle pulmonaire (HTAP - groupe 1). Une fois le diagnostic d'HTAP posé, l'accent est principalement mis sur l'importance de l'évaluation du risque et sur son application pour la stratégie thérapeutique et les besoins du patient.
Alors que les premières études sur l'HTAP étaient des études relativement brèves, à court terme, dont les critères d'évaluation primaires étaient des marqueurs de la capacité à l'effort - comme le test de marche de six minutes -, les études plus récentes sont plutôt des études à long terme qui utilisent un critère d'évaluation primaire composite de morbi-mortalité (SERAPHIN, GRIPHON, AMBITION, COMPASS-2). L'impact de la progression de la maladie a récemment été démontré dans les analyses décisives des études SERAPHIN et GRIPHON, dans lesquelles on a prouvé que la morbidité associée à l'HTAP, y compris les hospitalisations, constitue un solide facteur pronostique de mortalité. Ces données soulignent la nécessité d'une intensification précoce du traitement pour éviter une détérioration. Des registres récents qui utilisent la stratification du risque telle qu'elle est décrite dans les recommandations ESC/ERS de 2015 indiquent que l'obtention d'un profil à faible risque est associée au meilleur pronostic à long terme. Il est toutefois important de continuer à suivre les patients de manière stricte, étant donné que la plupart des patients présentant un profil à faible risque finiront quand même par développer une progression de la maladie.
De la stratification du risque à l'optimisation du traitement et du pronostic
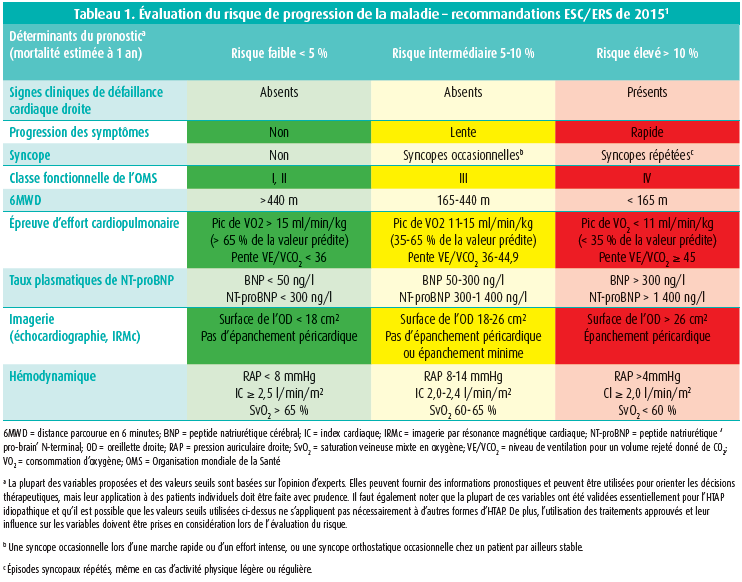
Une évaluation correcte du risque de progression de la maladie est capitale pour l'intensification du traitement de l'HTAP. La sévérité de l'HTAP doit être déterminée dès le diagnostic et par la suite, lors de chaque consultation de suivi. Les paramètres cliniques des patients peuvent être 'stables', même s'il y a une progression de la maladie, comme p. ex. une détérioration de la fonction ventriculaire droite. Il faut dès lors tenir compte de plusieurs indicateurs. Les limitations fonctionnelles et les modifications hémodynamiques sont les principaux déterminants de la sévérité de la maladie, et ils constituent dès lors la base de la stratification du risque telle qu'elle est décrite dans les recommandations ESC/ERS de 20151. Ces recommandations préconisent de déterminer la sévérité de l'HTAP à l'aide d'un ensemble de données provenant de l'évaluation clinique, des épreuves d'effort, des marqueurs biochimiques, de l'échocardiographie et des évaluations hémodynamiques (tableau 1 - recommandation de classe IC). Sur la base de ces résultats, on distingue un risque faible, intermédiaire et élevé, ce qui correspond à une mortalité estimée à 1 an de < 5 %, 5-10 % et > 10 %, respectivement. Il faut tendre à atteindre et conserver un profil à faible risque (recommandation de classe IC), ce qui est généralement associé à une bonne capacité à l'effort, une bonne qualité de vie, une bonne fonction ventriculaire droite et un risque de mortalité faible. Plus spécifiquement, cela revient à garder (ou amener) les patients en classe fonctionnelle II de l'OMS (OMS-FC). Les preuves étayant cette stratégie ont récemment été tirées de quelques registres européens2-4. Dans ces registres, le pronostic du patient est différencié sur la base du profil de risque au moment du diagnostic d'HTAP et lors du suivi. Les résultats indiquent que l'obtention ou le maintien d'un profil à faible risque sont associés à un net avantage en termes de survie2, 3. Ceci implique qu'un profil à risque intermédiaire doit être considéré comme une réponse inadéquate et que le traitement doit donc être optimisé le plus rapidement possible pour tendre à un profil à faible risque.
Depuis les recommandations ESC/ERS de 2015, l'instauration précoce d'une bithérapie est devenue la référence, en lieu et place d'une monothérapie. Dans la plupart des cas (OMS-FC II ou III), on commence par un antagoniste des récepteurs de l'endothéline (ERA) en combinaison avec un inhibiteur de la phosphodiestérase de type 5 (PDE-5i)1. Ceci est étayé par les résultats de l'étude AMBITION, qui indique que le traitement initial combinant de l'ambrisentan et du tadalafil améliore significativement l'évolution des patients en classes OMS-FC II et III5. Cette étude randomisée portant sur 500 patients souffrant d'HTAP nouvellement diagnostiquée, en classe OMS-FC II et III, compare la combinaison de 10 mg d'ambrisentan et de 40 mg de tadalafil avec chaque composant en monothérapie. Le critère d'évaluation primaire composite est la survenue du premier événement indiquant un échec clinique (défini comme un décès, une hospitalisation pour aggravation de l'HTAP, la progression de la maladie ou une réponse clinique insuffisante à long terme). Le traitement combiné administré pendant 18 mois en moyenne a été associé à une réduction de 50 % des échecs cliniques (18 % versus 31 %; p < 0,001). La réduction des échecs cliniques était essentiellement médiée par la diminution des hospitalisations dans le cadre de l'HTAP progressive, plutôt que par une amélioration de la survie ou de la classe OMSFC. Cette étude constitue la base de la recommandation de ce traitement combiné spécifique chez les patients souffrant d'HTAP, en classes OMS-FC II et III1. Chez les patients qui ont une contre-indication à un de ces agents, on donne la préférence à la substitution par un autre agent oral de la même classe, bien que ces combinaisons aient été moins étudiées et que des interactions médicamenteuses puissent potentiellement influencer l'évolution. L'étude SERAPHIN6 a démontré le bénéfice du macitentan. Dans cette étude, 742 patients souffrant d'HTAP ont été randomisés vers 3 mg ou 10 mg de macitentan, versus un placebo. La survenue du critère d'évaluation primaire composite (englobant décès, septostomie atriale, transplantation pulmonaire ou traitement par prostanoïdes parentéraux dans le cadre de la progression de la maladie) était réduite de 30 % (p = 0,01) et de 45 % (p < 0,001) dans les groupes macitentan 3 mg et 10 mg, respectivement, comparativement au groupe placebo. Ceci repose principalement sur la réduction de la progression de la maladie.
L'effet du traitement doit être strictement surveillé en prévoyant un suivi régulier tous les 3 à 6 mois (recommandation de classe IC). L'intensification du traitement, incluant l'association - à temps - d'un troisième agent, est recommandée chez la majorité des patients qui n'atteignent pas le statut à faible risque. Une trithérapie avec ajout de sélexipag - un agoniste oral sélectif des récepteurs à la prostacycline (agoniste des récepteurs IP) - à l'ERA et/ou au PDE5-i se voit attribuer une recommandation IB pour les patients en classes OMS-FC II et III et une recommandation IIA pour la classe OMS-FC IV. Cette recommandation est étayée entre autres par les résultats de l'étude GRIPHON, qui démontre un bénéfice significatif de l'association de sélexipag en plus de la bithérapie7. GRIPHON est une étude randomisée de phase 3, en double aveugle, dans laquelle 1 156 patients ont été randomisés vers sélexipag ou un placebo7. Il n'y a pas de différence significative entre les deux groupes en ce qui concerne le nombre de patients sous mono- ou bithérapie par ERA et/ou PDE-5i au moment de la randomisation. Le traitement par sélexipag, associé ou non à un ERA et/ou à un PDE-5i, entraîne une réduction de 40 % (p < 0,001) du critère d'évaluation primaire - un critère composite de morbi-mortalité (englobant mortalité, hospitalisation pour aggravation de l'HTAP, aggravation de l'HTAP nécessitant une transplantation pulmonaire ou une septostomie atriale, instauration de prostanoïdes parentéraux ou d'une oxygénothérapie chronique). Cette réduction du critère d'évaluation primaire est essentiellement médiée par la diminution de la progression de la maladie et des hospitalisations dans le groupe sélexipag. On n'a pas observé de différences significatives sur le plan de la mortalité entre les deux groupes.
Les résultats de GRIPHON et SERAPHIN soulignent en outre l'importance pronostique de la survenue d'une morbidité liée à l'HTAP6, 7. Les patients présentant un événement de morbidité (selon la définition des études GRIPHON et SERAPHIN) courent un risque de décès significativement plus élevé, qui persiste même après 12 mois de suivi. Ces résultats rappellent combien il est important d'éviter la progression de la maladie.
Conclusion
Ces dernières années, les preuves relatives à la prise en charge de l'HTAP se sont rapidement multipliées. Diverses classes thérapeutiques sont aujourd'hui disponibles. Il est dès lors essentiel d'optimiser l'utilisation de ces traitements. Une fois le diagnostic d'HTAP posé, une évaluation correcte du risque est capitale pour définir une stratégie thérapeutique individualisée.
Les progrès au niveau des options thérapeutiques pour l'HTAP ont débouché sur une amélioration du pronostic des patients. Le principal objectif du traitement de l'HTAP est d'éviter la morbidité, comme les hospitalisations, étant donné l'association décrite avec la mortalité. En instaurant ou en modifiant à temps un traitement combiné, il est possible de freiner la progression de l'HTAP. Par ailleurs, il est capital d'atteindre un profil à faible risque sous traitement. De vastes études ciblées sur les événements (event-driven), telles que GRIPHON et SERAPHIN, ont démontré que la combinaison des traitements de l'HTAP peut améliorer le pronostic à long terme des patients.
Références
- Galie, N., Humbert, M., Vachiery, J.L., Gibbs, S., Lang, I., Torbicki, A., et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J, 2016, 37 (1), 67-119.
- Kylhammar, D., Kjellstrom, B., Hjalmarsson, C., Jansson, K., Nisell, M., Soderberg, S., et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J, 2017, doi: 10.1093/eurheartj/ehx257. [Epub ahead of print].
- Hoeper, M.M., Kramer, T., Pan, Z., Eichstaedt, C.A., Spiesshoefer, J., Benjamin, N., et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J, 2017, 50 (2), 1700740; DOI: 10.1183/13993003.00740- 2017.
- Boucly, A., Weatherald, J., Savale, L., Jais, X., Cottin, V., Prevot, G., et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J, 2017, 50 (2), pii: 1700889. doi: 10.1183/13993003.00889-2017
- Galie, N., Barbera, J.A., Frost, A.E., Ghofrani, H.A., Hoeper, M.M., McLaughlin, V.V., et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373 (9), 834-844.
- Pulido, T., Adzerikho, I., Channick, R.N., Delcroix, M., Galie, N., Ghofrani, H.A., et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med, 2013, 369 (9), 809-918.
- Sitbon, O., Channick, R., Chin, K.M., Frey, A., Gaine, S., Galie, N., et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373 (26), 2522-2533.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.