Focus on clinical management of pulmonary arterial hypertension. ESC Congress session 2018.
Le diagnostic d'hypertension pulmonaire (HTP) repose sur une définition hémodynamique avec une pression artérielle pulmonaire moyenne (PAPm) mesurée au cathétérisme cardiaque droit ≥ 25 mmHg au repos (valeur normale: 14 +/-3 mmHg). Aussi la mesure de la pression artérielle pulmonaire occluse (PAPO) permet la distinction entre une origine pré-capillaire (PAPO ≤ 15 mmHg) et post-capillaire (PAPO > 15 mmHg). L'hypertension artérielle pulmonaire se définit par l'association de critères hémodynamiques (PAPm ≥ 25 mmHg , PAPO ≤ 15 mmHg et résistances vasculaires pulmonaires (RVP) > 3UW) et cliniques (absence de conditions cliniques compatibles avec un autre groupe d'hypertension pulmonaire pré-capillaire).
Pulmonary arterial hypertension diagnosis, a challenge
L'hypertension artérielle pulmonaire (HTAP) rassemble plusieurs formes dont les plus fréquentes sont par ordre décroissant l'idiopathique, les connectivites (sclérose systémique) et les cardiopathies congénitales (shunts systémico-pulmonaires) avec la caractéristique histologique commune d'un même remodelage du lit artériel pulmonaire (lésions plexiformes dues à la prolifération de cellules endothéliales et musculaires lisses).
Néanmoins, le diagnostic reste complexe pour plusieurs raisons:
- une faible prévalence (15-60/1,106 adultes) et incidence (5-10/1,106 par an);
- un âge plus avancé au moment du diagnostic rendant plus complexe le diagnostic de certitude au vu des comorbidités plus fréquentes (obésité, hypertension artérielle, pathologie pulmonaires associées, etc.);
- de multiples causes potentielles d'HTP;
- l'absence de présentation clinique spécifique (dyspnée à l'effort, oedème, fatigue, douleur thoracique) et chevauchement avec d'autres pathologies (asthme, pathologie du coeur gauche, bronchite chronique).
La reconnaissance de l'hypertension pulmonaire et notamment de l'HTAP est donc complexe parmi des patients plus âgés avec de multiples comorbidités. Dès lors le diagnostic est souvent posé à un stade avancé de la pathologie (patients en classe fonctionnelle III ou IV) et en retard par rapport à l'apparition des symptômes. En référence aux travaux de Brown et al.3 issus du registre REVEAL, un âge jeune < 36 ans et la présence d'une affection pulmonaire concomitante (trouble ventilatoire obstructif/ syndrome des apnées du sommeil) participent à retarder le diagnostic (> 2 ans). Aussi, la réalisation d'un cathétérisme droit dans un centre de référence est une étape-clé dans la prise en charge du patient d'autant plus pour les formes pré-capillaires comme l'HTAP pour laquelle il existe un traitement spécifique.
Assessing the risk of mortality and disease progression
Le management de l'HTAP représente un défi clinique majeur à la fois pour poser un diagnostic précis et précoce mais aussi pour instaurer un traitement spécifique à temps. Trop souvent tardif, le diagnostic à un stade avancé et souvent irréversible s'associe à un pronostic extrêmement sévère (50 % à 5 ans pour des patients en classe fonctionnelle III-IV)5. Comme le traitement a un impact sur la morbidité et la mortalité, il est d'autant plus important d'instaurer un traitement le plus tôt possible.
Citons par exemple, une sous-analyse de l'étude GRIPHON6 portant sur des patients atteints de sclérose systémique dont on connaît la difficulté thérapeutique et qui a démontré un bénéfice de l'utilisation d'un agoniste sélectif du récepteur de la prostacycline (selexipag) en termes de réduction de progression de l'HTAP (réduction du risque de 41 %; (HR 0,59; 95 % CI: 0,41-0,85) par rapport au placebo. L'intérêt d'une triple thérapie pour impacter davantage le pronostic ne cesse de prendre de l'ampleur comme en témoignent les travaux de Coghlan et al.4 ayant mis en évidence la valeur ajoutée du selexipag associé à une double thérapie (ERA + PDE-5i) sur la réduction du risque de progression de l'HTAP pour des patients à risque intermédiaire (WHO II-III) (figure 1).
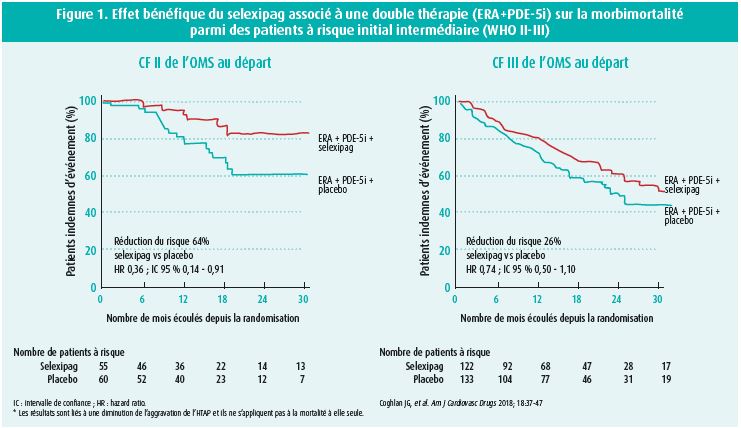
L'approche basée sur le profil de risque du patient (faible/intermédiaire/haut risque) est une pierre angulaire dans la prise en charge multimodale de l'HTAP par des centres experts1. La classification repose sur une analyse multiparamétrique clinique (classe fonctionnelle, présence de signes de gravité comme la syncope, capacité physique VO2), imagerie (fonction ventriculaire droite), hémodynamiques (pression oreillette droite, index cardiaque et saturation veineuse en oxygène) et biochimiques (NT-pro BNP). Plus le patient remplit les critères du profil à faible risque de mortalité, meilleure en est sa survie et ce au moment du diagnostic mais surtout lors du suivi endéans l'année du diagnostic2 (figure 2).
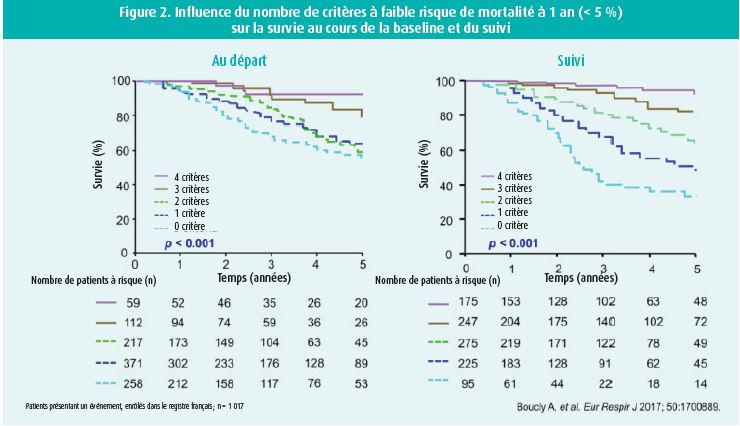
Le design des études relatives à l'HTAP ne cesse d'évoluer avec l'identification de puissants marqueurs pronostiques. Prenons notamment l'exemple de l'amélioration du test de marche de 6 min initialement utilisé comme objectif clinique d'un traitement efficace, bien que facilement réalisable, ne reflète pas le bénéfice clinique thérapeutique11.
En revanche, les données issues des registres incriminent la progression de l'HTAP comme étant fortement associée à un mauvais pronostic10. Le concept de progression de l'HTAP regroupe la survenue de morbidités telles qu'une aggravation des symptômes et de l'incapacité fonctionnelle mais aussi une hospitalisation, une transplantation pulmonaire, etc.). Mentionnons les travaux de Mclaughlin et al8. dérivés des études SERAPHIN et GRIPHON à travers lesquels, la survenue d'un évènement morbide dans les 3 mois de la randomisation était associée à un risque accru de mortalité (SERAPHIN (HR: 3,39; 95 % CI: 1,94-5,92); GRIPHON (HR: 4,48; 95 % CI: 2,98-6,73). Dès lors, c'est en priorité la morbi-mortalité à long terme qui est évaluée pour juger de l'efficacité des traitements11.
Therapeutics strategies for pulmonary arteriel hypertension
La supériorité d'une double thérapie par rapport à une monothérapie est aujourd'hui bien établie mais ne suffit pas toujours à amener le patient dans une catégorie à faible risque de mortalité (< 5 %)1. Dans l'optique d'optimiser les stratégies thérapeutiques, la communauté scientifique concentre son intérêt sur la triple thérapie qui cible simultanément les 3 voies métaboliques (voies de l'endothéline (A), de l'oxyde nitrique (B) et de la prostacycline (C)) en vue d'une synergie des bénéfices.
En référence aux travaux de Sitbon et al.14, l'instauration d'une triple thérapie initiale (bosentan (A) + sildenafil (B) + epoprostenol (C)) pour les patients à haut risque de mortalité (WHO III-IV) a montré des bénéfices à long terme tant hémodynamiques que fonctionnels mais également en terme de survie.
à travers l'essai randomisé GRIPHON12-13, la triple thérapie (agoniste sélectif du récepteur de la prostacycline (selexipag) + double thérapie (ERA; PDE-5i) vs placebo + double thérapie (ERA;PDE-5I) cette fois séquentielle s'affiche supérieure à une double thérapie (ERA; PDE-5i) avec une réduction du risque d'évènements morbides de 37 % (HR 0,63, 99 % CI 0,39-1,01) pour les patients à risque intermédiaire (WHO II-III). Il n'y avait cependant pas de différence statistiquement significative en termes de mortalité entre les deux groupes.
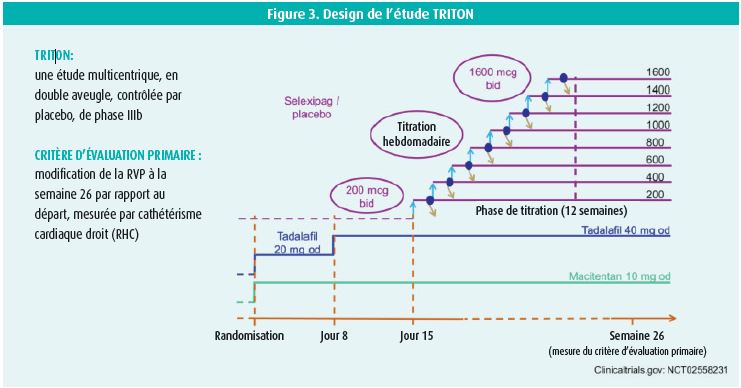
L'étude TRITON (figure 3), dont les résultats sont fort attendus, s'intéresse à la valeur ajoutée en première intention d'une triple thérapie orale initiale (agoniste sélectif du récepteur de la prostacycline (selexipag) + ERA (macitentan) + PDE-5i (tadalafil)) par rapport à une double thérapie (macitentan + tadalafil + placebo).
En résumé, cette session a tout d'abord permis de rappeler l'importance pour le clinicien d'être alerte au diagnostic d'hypertension artérielle pulmonaire face à une symptomatologie peu spécifique puis de poser un diagnostic précis et précoce en vue d'une stratégie thérapeutique basée sur le profil de risque du patient. La triple thérapie prend de plus en plus d'essor dans l'algorithme décisionnel. Ensuite, gardons à l'esprit que la dysfonction ventriculaire droite est le principal facteur qui péjore le pronostic d'où l'importance de reconnaître précocement l'HTAP pour débuter un traitement qui a un impact notable sur la survie. Enfin, la réévaluation régulière de la stabilité de la maladie est essentielle pour adapter la thérapeutique et pour prévenir les comorbidités qui assombrissent le pronostic de l'HTAP.
Références
- Galie, N., Humbert, M., Vachiery, J.L., et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Resp J, 2015, 46, 903-975.
- Badesch, D.B., Raskob, G.E., Elliott, C.G., et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest, 2010, 137, 376-387.
- Brown, L.M., Chen, H., Halpern, S., et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL Registry. Chest, 2011, 140, 19-26.
- Coghlan, J.G., Channick, R., Chin, K., et al. Targeting the Prostacyclin Pathway with Selexipag in Patients with Pulmonary Arterial Hypertension Receiving Double Combination Therapy: Insights from the Randomized Controlled GRIPHON Study. Am J Cardiovasc Drugs, 2018, 18, 37-47.
- Farber, H.W., Miller, D.P., Poms, A.D., et al. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest, 2015, 148, 1043-1054.
- Gaine, S., Chin, K., Coghlan, G., et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Resp J, 2017, 18, 37-47.
- Gaine, S., McLaughlin, V. Pulmonary arterial hypertension: tailoring treatment to risk in the current era. Eur Resp Rev, 2017, DOI: 10.1183/16000617.0095-2017.
- McLaughlin, V.V., Hoeper, M.M., Channick, R.N., et al. Pulmonary Arterial Hypertension- Related Morbidity Is Prognostic for Mortality. J Am Coll Cardiol, 2018, 71, 752-763.
- Mehta, S., Vachiery, J.L. Pulmonary hypertension: the importance of correctly diagnosing the cause. Eur Resp Rev, 2016, 25, 372-380.
- Preston, I.R., Suissa, S., Humbert, M. New perspectives in long-term outcomes in clinical trials of pulmonary arterial hypertension. Eur Resp Rev, 2013, 22, 495-502.
- Savarese, G., Paolillo, S., Costanzo, P., et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol, 2012, 60, 1192-1201.
- Sitbon, O., Channick, R., Chin, K.M., et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373, 2522-2533.
- Sitbon, O., Gaine, S. Beyond a single pathway: combination therapy in pulmonary arterial hypertension. Eur Resp Rev, 2016, 25, 408-417.
- Sitbon, O., Jais, X., Savale, L., et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Resp J, 2014, 43, 1691-1697.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.