Focus on clinical management of pulmonary arterial hypertension. ESC Congress session 2018.
De diagnose van pulmonale arteriële hypertensie (PAH) berust op een hemodynamische definitie met een gemiddelde pulmonale arteriële druk (PAPm) gemeten met rechterhartkatheterisatie ≥ 25 mmHg in rust (normale waarde: 14 +/-3 mmHg). Met een meting van de pulmonale arteriële druk onder occlusie (PAPO) kan het onderscheid worden gemaakt tussen een precapillaire oorsprong (PAPO ≤ 15 mmHg) en een postcapillaire oorsprong (PAPO > 15 mmHg). Pulmonale hypertensie wordt gedefinieerd als een combinatie van hemodynamische criteria (PAPm ≥ 25 mmHg, PAPO ≤ 15 mmHg en pulmonale vaatweerstand (PVR) > 3WU) en klinische criteria (geen klinische tekenen die verenigbaar zijn met een andere groep van precapillaire pulmonale hypertensie).
Pulmonary arterial hypertension diagnosis, a challenge
Er bestaan meerdere vormen van pulmonale arteriële hypertensie (PAH), met in dalende volgorde van frequentie de idiopathische vorm, de systemische vorm (systeemsclerose) en congenitaal hartlijden (systemisch-pulmonale shunts) met als gemeenschappelijk histologisch kenmerk een remodeling van het pulmonale arteriële vaatbed (plexiforme letsels door proliferatie van endotheelcellen en gladde spiercellen).
Toch blijft de diagnose om meerdere redenen complex:
- een lage prevalentie (15-60/1,106 volwassenen) en incidentie (5-10/1,106 per jaar);
- een hogere leeftijd op het moment van de diagnose, waardoor het moeilijker is om een zekerheidsdiagnose te stellen omdat er vaker comorbiditeiten aanwezig zijn (obesitas, hypertensie, begeleidende longaandoeningen, enz.);
- de verschillende mogelijke oorzaken van PAH;
- het ontbreken van een specifiek klinisch beeld (dyspneu bij inspanning, oedeem, vermoeidheid, pijn op de borst) en overlapping met andere aandoeningen (astma, linkerhartziekte, chronische bronchitis).
Bij oudere patiënten met meerdere comorbiditeiten is het dan ook moeilijk om pulmonale hypertensie en met name PAH te herkennen. Daardoor wordt de diagnose vaak in een laat stadium van de ziekte gesteld (patiënten in functionele klasse III of IV) en niet op het moment dat de symptomen verschijnen. Uit het onderzoek van Brown et al.3 op basis van het REVEAL-register blijkt dat een jonge leeftijd < 36 jaar en de aanwezigheid van een begeleidende longaandoening (obstructieve ademhalingsstoornis/ slaapapneusyndroom) ertoe bijdragen dat de diagnose pas later gesteld wordt (> 2 jaar). De uitvoering van een rechterhartkatheterisatie in een referentiecentrum is dan ook een cruciale stap in de aanpak van de patiënt, zeker voor precapillaire vormen van PAH waarvoor een specifieke behandeling bestaat.
Assessing the risk of mortality and disease progression
Het beleid bij PAH vormt een grote uitdaging op klinisch vlak, zowel om vroegtijdige een precieze diagnose te stellen als om tijdig een specifieke behandeling te kunnen geven. Al te vaak wordt de diagnose te laat gesteld, in een gevorderd en vaak onomkeerbaar stadium, en in dat geval is de prognose uiterst somber (5 jaarsoverleving van 50 % voor patiënten in de functionele klassen III-IV)5. Aangezien de behandeling een invloed heeft op de morbiditeit en de mortaliteit, is het des te belangrijker om patiënten zo vroeg mogelijk te behandelen.
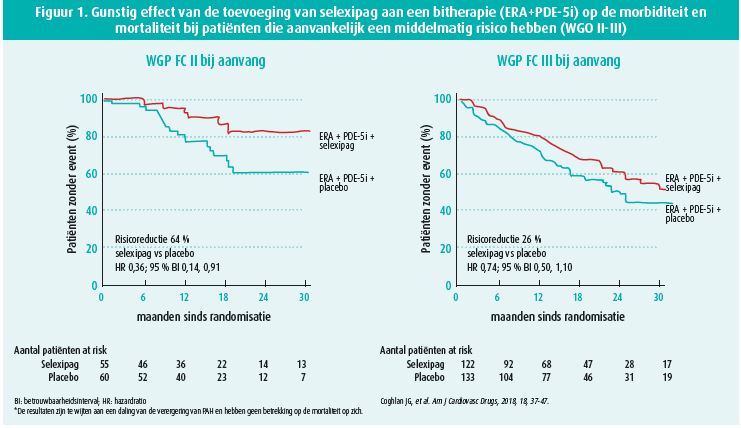
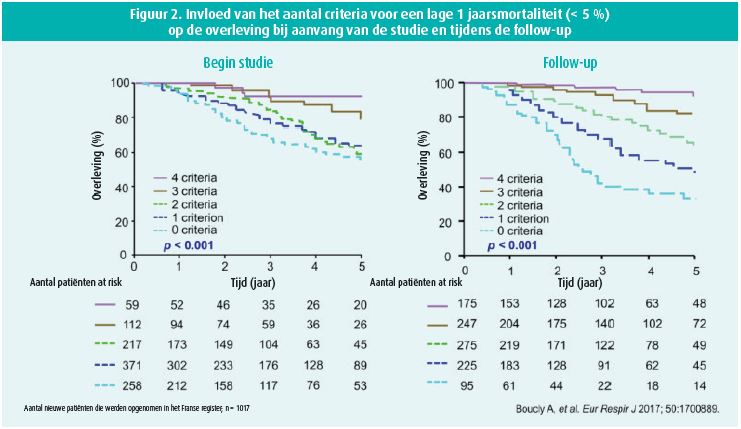
We verwijzen daarvoor naar een subanalyse van de GRIPHON-studie6. Die werd uitgevoerd bij patiënten met systemische sclerose, wat erg moeilijk te behandelen is. Uit de studie is gebleken dat het gebruik van een selectieve prostacyclinereceptoragonist (selexipag) de progressie van PAH vertraagt (daling van het risico met 41 %; (HR 0,59; 95 % BI: 0,41-0,85) in vergelijking met een placebo. Er zijn almaar meer bewijzen dat een triple-therapie de prognose kan verbeteren, en dat blijkt ook uit de studies van Coghlan et al.4, waarin werd aangetoond dat toevoeging van selexipag aan een bitherapie (ERA + PDE-5i) het risico op progressie van PAH verlaagt bij patiënten met een middelmatig risico (WGO II-III) (figuur 1). In expertcentra vormt een benadering die gebaseerd is op het risicoprofiel van de patiënt (laag/middelmatig/hoog risico) de hoeksteen van de multimodale aanpak van PAH1. De classificatie berust op een analyse van meerdere klinische parameters (functionele klasse, aanwezigheid van tekenen van ernst zoals syncope, lichamelijke conditie, VO2), beeldvormingsonderzoek (rechterventrikelfunctie), hemodynamische parameters (druk in rechterboezem, cardiale index en veneuze zuurstofsaturatie) en biochemische parameters (NT-pro BNP). Hoe beter de patiënt voldoet aan de criteria van het profiel met een lager mortaliteitsrisico, hoe beter zijn overleving. Dat geldt op het moment van de diagnose, maar vooral tijdens de follow-up binnen een jaar na de diagnose2 (figuur 2).
Het opzet van de studies naar PAH evolueert voortdurend aangezien er nu krachtige prognostische markers bekend zijn. Zo werd de 6 minutenlooptest, die gemakkelijk uit te voeren is, vroeger gebruikt als klinische doelstelling voor een werkzame behandeling, maar geeft die het klinische voordeel van de behandeling niet correct weer11.
Anderzijds is uit de gegevens van de registers gebleken dat er een sterke correlatie bestaat tussen de progressie van de PAH en een slechte prognose10. Progressie van PAH wordt gedefinieerd als een geheel van fenomenen waaronder een verergering van de symptomen en van de longfunctie, maar ook een opname in het ziekenhuis, een longtransplantatie, enz. Mclaughlin et al8. hebben op basis van de gegevens van de studies SERAPHIN en GRIPHON aangetoond dat het optreden van een morbiditeit binnen 3 maanden na randomisatie gepaard ging met een hogere mortaliteit (SERAPHIN (HR: 3,39; 95 % BI: 1,94-5,92); GRIPHON (HR: 4,48; 95 % BI: 2,98-6,73). Om de werkzaamheid van de behandelingen te beoordelen, moeten we dus vooral kijken naar de morbiditeit en de mortaliteit op lange termijn11.
Therapeutics strategies for pulmonary arteriel hypertension
Er is ondertussen duidelijk aangetoond dat een bitherapie betere resultaten geeft dan een monotherapie, maar ook daarmee komt de patiënt niet altijd in een categorie met een lage mortaliteit (< 5 %)1 terecht. Om de behandelingsstrategie te optimaliseren, concentreert de wetenschappelijke gemeenschap haar inspanningen op de tripeltherapie, die op de 3 metabole wegen tegelijk ingrijpt (de endothelinepathway (A), de stikstofoxidepathway (B) en de prostacyclinepathway (C)) om tot een synergetische werking te komen.
Uit het onderzoek van Sitbon et al.14 is gebleken dat meteen een tripeltherapie (bosentan (A) + sildenafil (B) + epoprostenol (C)) geven aan patiënten met een hoog mortaliteitsrisico (WGO III-IV) op lange termijn zowel hemodynamische als functionele voordelen biedt, en ook de overleving verbetert.
In de gerandomiseerde studie GRIPHON12-13 werden met een sequentiële tripeltherapie (selectieve prostacyclinereceptoragonist (selexipag) + bitherapie (ERA; PDE-5i) betere resultaten behaald dan met een placebo + bitherapie (ERA; PDE-5I). Het risico op morbide gebeurtenissen daalde met 37 % (HR 0,63, 99 % BI 0,39-1,01) bij patiënten met een middelmatig risico (WGO II-III). De mortaliteit was echter niet significant verschillend tussen beide groepen.
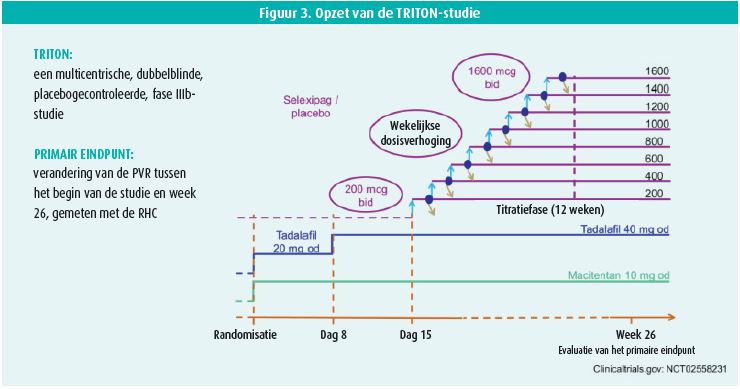
De TRITON-studie (figuur 3) onderzoekt de toegevoegde waarde van een initiële orale tripeltherapie (selectieve prostacyclinereceptoragonist (selexipag) + ERA (macitentan) + PDE-5i (tadalafil)) ten opzichte van een bitherapie (macitentan + tadalafil + placebo). Naar de resultaten van die studie wordt met ongeduld uitgekeken.
Samenvattend kunnen we stellen dat deze sessie de clinicus eraan heeft herinnerd dat het belangrijk is om aan de diagnose van arteriële pulmonale hypertensie te denken bij een weinig specifieke symptomatologie, zodat snel een precieze diagnose kan worden gesteld en een behandeling kan worden gestart op basis van het risicoprofiel van de patiënt. De tripeltherapie krijgt in de beslissingsboom een almaar centralere rol toebedeeld. Verder mogen we niet vergeten dat een rechterventrikeldisfunctie de belangrijkste factor is voor een slechte prognose. Het is dan ook belangrijk om PAH vroeg te herkennen, zodat een behandeling kan worden gestart die de overleving in belangrijke mate verbetert. Tot slot moet regelmatig gecontroleerd worden of de ziekte stabiel is, zodat de behandeling kan worden aangepast en morbiditeiten die de prognose van PAH verslechteren kunnen worden voorkomen.
Referenties
- Galie, N., Humbert, M., Vachiery, J.L., et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Resp J, 2015, 46, 903-975.
- Badesch, D.B., Raskob, G.E., Elliott, C.G., et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest, 2010, 137, 376-387.
- Brown, L.M., Chen, H., Halpern, S., et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL Registry. Chest, 2011, 140, 19-26.
- Coghlan, J.G., Channick, R., Chin, K., et al. Targeting the Prostacyclin Pathway with Selexipag in Patients with Pulmonary Arterial Hypertension Receiving Double Combination Therapy: Insights from the Randomized Controlled GRIPHON Study. Am J Cardiovasc Drugs, 2018, 18, 37-47.
- Farber, H.W., Miller, D.P., Poms, A.D., et al. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest, 2015, 148, 1043-1054.
- Gaine, S., Chin, K., Coghlan, G., et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Resp J, 2017, 18, 37-47.
- Gaine, S., McLaughlin, V. Pulmonary arterial hypertension: tailoring treatment to risk in the current era. Eur Resp Rev, 2017, DOI: 10.1183/16000617.0095-2017.
- McLaughlin, V.V., Hoeper, M.M., Channick, R.N., et al. Pulmonary Arterial Hypertension- Related Morbidity Is Prognostic for Mortality. J Am Coll Cardiol, 2018, 71, 752-763.
- Mehta, S., Vachiery, J.L. Pulmonary hypertension: the importance of correctly diagnosing the cause. Eur Resp Rev, 2016, 25, 372-380.
- Preston, I.R., Suissa, S., Humbert, M. New perspectives in long-term outcomes in clinical trials of pulmonary arterial hypertension. Eur Resp Rev, 2013, 22, 495-502.
- Savarese, G., Paolillo, S., Costanzo, P., et al. Do changes of 6-minute walk distance predict clinical events in patients with pulmonary arterial hypertension? A meta-analysis of 22 randomized trials. J Am Coll Cardiol, 2012, 60, 1192-1201.
- Sitbon, O., Channick, R., Chin, K.M., et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med, 2015, 373, 2522-2533.
- Sitbon, O., Gaine, S. Beyond a single pathway: combination therapy in pulmonary arterial hypertension. Eur Resp Rev, 2016, 25, 408-417.
- Sitbon, O., Jais, X., Savale, L., et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Resp J, 2014, 43, 1691-1697.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.