Congrès de l'ESC 2020
Compte rendu du symposium
Informations générales
L'amylose cardiaque est une cardiomyopathie infiltrative due au dépôt extracellulaire progressif de fibrilles d'amyloïde insolubles, formées à partir de protéines précurseurs. Il s'agit le plus souvent d'une affection systémique dans laquelle d'autres systèmes d'organes peuvent être infiltrés, comme le système neural, oculaire, rénal, gastro-intestinal et musculo-squelettique, ce qui se traduit par des dysfonctions organiques symptomatiques. Deux protéines précurseurs causent 95 % des cas d'amylose cardiaque : soit il s'agit de chaînes légères d'immunoglobuline-G (« amyloïdose lightchain » ou « AL ») soit, plus fréquemment, de la transthyrétine (ATTR), une protéine de transport sérique1. La cardiomyopathie amyloïde de type AL est une tumeur hématologique due à la surproduction et au dépôt monoclonaux de chaînes légères d'immunoglobulines. La cardiomyopathie amyloïde à transthyrétine (ATTR-CMP) existe sous deux formes. L'ATTR de type sauvage (« wildtype ») résulte de la dissociation idiopathique de la protéine transthyrétine normale en monomères de fibrilles d'amyloïde chez, principalement, les hommes caucasiens (9:1) âgés de 70 ans et plus (un âge plus jeune ou plus avancé est possible). Il s'agit de la forme la plus fréquente d'amylose cardiaque, qui survient notamment chez 13 % des personnes âgées présentant une insuffisance cardiaque et une fraction d'éjection conservée (HFpEF), chez 16 % des patients ayant subi une implantation percutanée d'une valve aortique (TAVI) et chez 6 % des patients atteints de cardiomyopathie hypertrophique1. L'ATTR mutée est la conséquence d'une configuration anormale de la protéine transthyrétine du fait d'une mutation génétique (> 120 mutations ont été décrites) au niveau du gène TTR ; elle s'observe chez les hommes comme chez les femmes à partir de l'âge de 40-50 ans (un âge plus jeune ou plus avancé est possible) et est associée à une certaine prévalence géographique et ethnique. La distinction entre ces trois formes d'amylose cardiaque est essentielle car les pronostics et les traitements qui y sont associés varient fortement selon la forme. Après un diagnostic d'amylose cardiaque symptomatique, la durée de survie moyenne est de respectivement 6 mois (AL), 3,5 ans (ATTR de type sauvage) et 2-5 ans (ATTR mutée)1. La distinction entre les différentes formes repose sur l'intégration du profil clinique, d'analyses sanguines et d'examens d'imagerie cardiaque non invasifs, parfois complétés par une biopsie tissulaire. Il est toutefois important de noter qu'aucun examen d'imagerie ne permet d'opérer la distinction définitive. En raison de la connaissance limitée de la maladie, celleci fait l'objet de diagnostics erronés dans 35 % des cas. Or, un diagnostic précoce est essentiel pour proposer au patient une chimio- et/ou immunothérapie en temps opportun (dans le cas de l'AL), ou encore une stabilisation des tétramères et/ou le silençage du gène TTR (dans le cas de l'ATTR)1, 2.
Quand suspecter la présence d'une ATTR-CMP ?
L'infiltration de l'amyloïde dans plusieurs systèmes organiques induit des modifications structurelles et fonctionnelles dans les différents organes, ce qui se traduit par un ensemble de symptômes/signes cliniques typiques ou suspects, et d'anomalies à l'imagerie cardiaque (tableau 1).
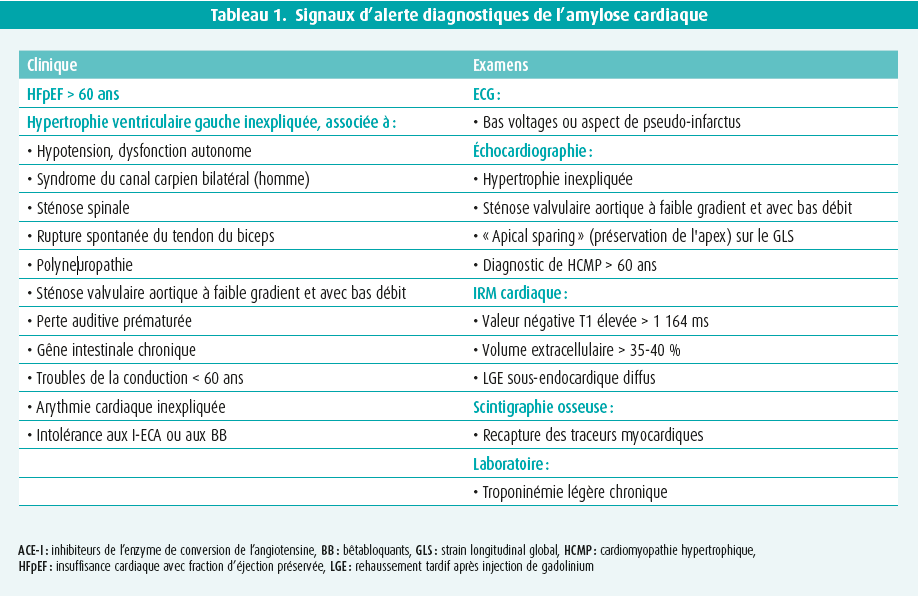
Signaux d'alerte cliniques
La clinique cardiaque comprend principalement le tableau HFpEF, qui apparaît très souvent comme premier signe et qui sera particulièrement suspect chez les hommes âgés de plus de 65 ans. Ce tableau est presque toujours associé à une élévation du NT-proBNP et à une légère troponinémie. Par ailleurs, la survenue d'une arythmie cardiaque et/ou de troubles de la conduction inexpliqués (détresse du stimulateur cardiaque), surtout à un jeune âge, est également suspecte. L'ecg est aspécifique, mais suspect en cas de bas voltages et d'aspect de pseudo-infarctus. Un angor, une claudication de la mâchoire et une ischémie secondaire à une infiltration (micro)vasculaire, ainsi qu'une thrombo-embolie inexpliquée, sont possibles1.
Des symptômes ou signes extracardiaques d'infiltration progressive coexistent fréquemment, souvent cinq à dix ans avant le diagnostic1. Dans ce contexte clinique, des antécédents de syndrome de canal carpien (bilatéral, surtout chez les hommes), sténose spinale, rupture spontanée du tendon du biceps, doigt à ressaut, dysfonctionnement autonome (hypotension, gêne gastro-intestinale inexpliquée, perte de poids), perte auditive et polyneuropathie sensorielle périphérique et/ou motrice inexpliquée, sont extrêmement suspects. Un glaucome peut également apparaître. Dans l'ATTR mutée, indépendamment du type de mutation, un phénotype clinique de neuropathie ou de cardiomyopathie, ou encore une forme mixte, est prépondérant et peut se manifester dès la trentaine ou la quarantaine. Les signes plus spécifiques de l'AL sont le purpura périorbitaire, l'éruption cutanée et la macroglossie (10 %).
Imagerie cardiaque suspecte
L'échocardiographie montre une hypertrophie allant d'une forme légère à prononcée du ventricule gauche (et droit), avec un aspect granulaire. Bien que généralement concentrique, cette hypertrophie peut être légèrement asymétrique et s'accompagner ou non d'une obstruction de la chambre de chasse. La fraction d'éjection du ventricule gauche reste le plus souvent préservée jusqu'à un stade avancé de la maladie, tandis qu'une dysfonction longitudinale modérée à prononcée est souvent déjà présente dès le stade précoce. Cette dysfonction est typique des segments basaux et moyens, mais pas des segments apicaux, ce qui génère le tableau quasi pathognomonique de l'apical sparing (préservation de l'apex) sur le « global longitudinal strain bull's eye plot » (figure 1). La présence d'une dysfonction diastolique, de signes de pressions de remplissage accrues et d'une dilatation biauriculaire est fréquente. On observe parfois un épanchement péricardique léger et un épaississement des lames avec une légère fuite1.
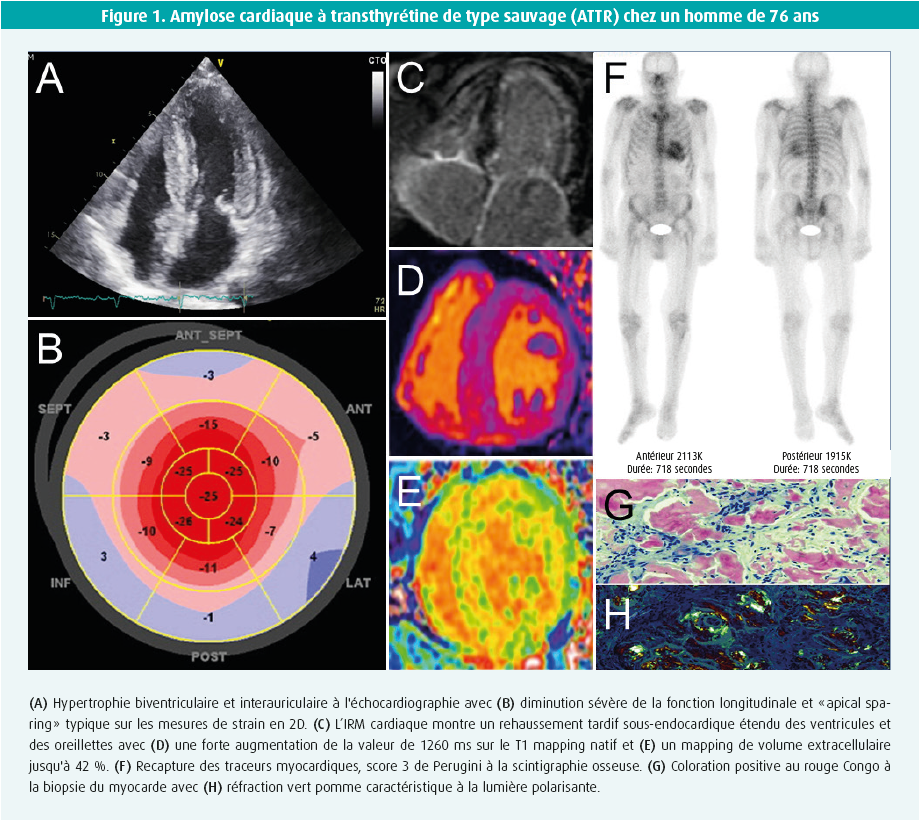
Outre le diagnostic de l'hypertrophie ventriculaire, l'IRM cardiaque offre surtout l'avantage de permettre la caractérisation tissulaire sur la base du rehaussement tardif après injection de gadolinium (late gadolinium enhancement, ou LGE, degré de fibrose focale) et de déterminer le volume extracellulaire (ECV, degré de la fibrose diffuse). Un LGE est généralement présent, parfois sous la forme d'une couche sous-endocardique diffuse (pathognomonique), mais plus fréquemment sous la forme d'un schéma plutôt tacheté ou transmural. L'ECV est généralement fortement accru, soit > 35-40 % (normal < 25 %) (figure 1). Par ailleurs, les éléments suivants sont relativement typiques : temps de relaxation élevé T1 (sans contraste), annulation difficile pour tout un intervalle de temps d'inversion, pool sanguin plus sombre et suppression anormale/difficile d'un myocarde infiltré souvent diffus1, 2.
Diagnostic pratique de l'ATTR-CMP
En cas de suspicion d'amylose cardiaque sur la base des signaux d'alerte cliniques, de l'échocardiographie, de l'IRM cardiaque et/ou de biomarqueurs, on procèdera au diagnostic par étapes2 (figure 2).
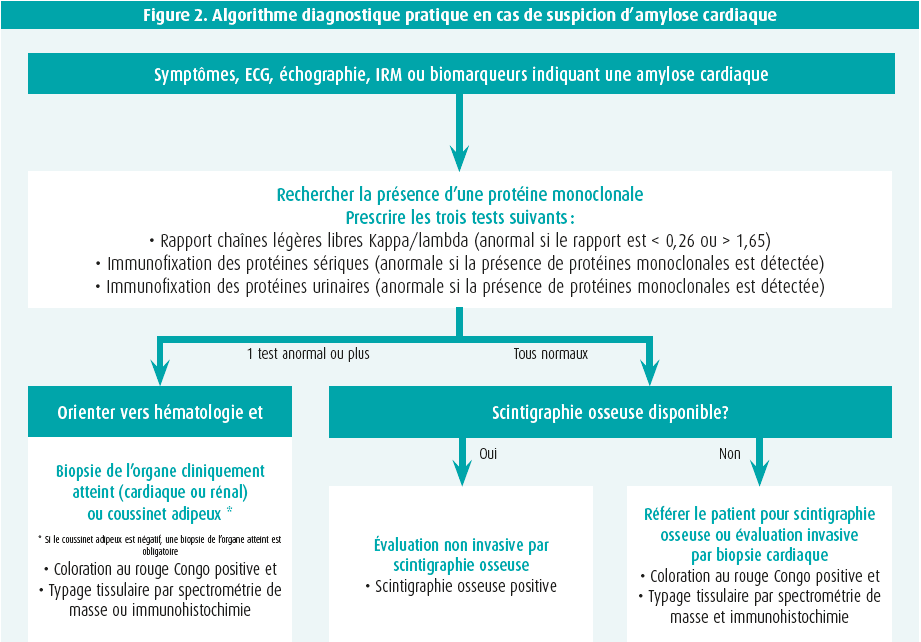
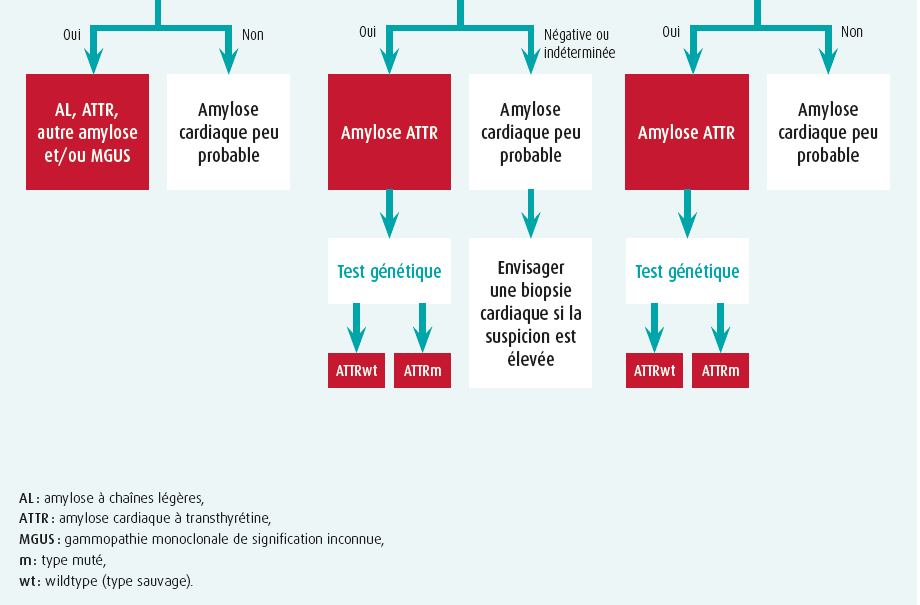
étape 1 : exclusion de la monoclonalité
Trois tests doivent être réalisés : un échantillon sanguin et un échantillon d'urine, chacun avec électrophorèse des protéines et immunofixation (immunoglobulines anormales et monoclonales), et dosage sérique des chaînes légères libres (anormal si le rapport kappa-lambda est < 0,26 ou > 1,65). Un avis hématologique est indiqué lorsque l'un de ces trois tests est anormal, une biopsie de la moelle osseuse ainsi qu'une biopsie d'organe ciblée (cardiaque ou rénale) pouvant alors s'avérer nécessaires. Attention : une gammopathie monoclonale de signification inconnue (MGUS) survient chez 3,2 % des patients de plus de 50 ans et est donc fréquemment concomitante de l'ATTR2.
étape 2 : scintigraphie osseuse
Les traceurs osseux marqués au technétium possèdent une affinité (qui n'est pas entièrement expliquée) pour, principalement, la TTR-amyloïde et parfois (affinité plutôt faible) pour l'AL. L'étape suivante consiste donc en une scintigraphie osseuse-SPECT évaluant la recapture myocardique, ainsi qu'en des coupes tomographiques. Soit on n'observe aucune recapture des traceurs myocardiques (score 0 de perugini), soit on observe une recapture qui est comparée avec le dessin du squelette environnant : score 1 de perugini (moins que le squelette), score 2 (équivalent au squelette) ou score 3 (plus que le squelette). En l'absence de monoclonalité, une recapture de score 2 ou plus présente une valeur prédictive de 100 % pour l'ATTR, la biopsie éventuelle du myocarde n'étant alors plus requise1, 2 (figure 1). Une recapture de score 0 ou 1 est toutefois possible avec l'AL ou avec l'ATTR (de stade précoce) et requiert une biopsie organique. Différents traceurs de PET-scan se sont révélés prometteurs pour le diagnostic de l'amylose, mais ils sont aujourd'hui principalement utilisés à des fins de recherche.
étape 3 : confirmation de l'amylose cardiaque
La présence d'une ATTR est confirmée lorsque la recapture des traceurs myocardiques sans monoclonalité est de score 2 ou plus. Les autres situations requièrent une biopsie. Une biopsie de graisse est facile à pratiquer, mais possède une sensibilité limitée (AL 84 %, ATTR mutée 45 % et ATTR type sauvage seulement 15 %)1. Une biopsie ciblée d'un organe atteint (rénal ou cardiaque) est donc nettement plus sensible. L'infiltration par l'amyloïde cardiaque n'est pas toujours homogène et l'IRM cardiaque avec LGE peut orienter le professionnel, lui permettant d'éviter l'erreur d'échantillonnage (résultat faux négatif). L'amyloïde se colore après une coloration au rouge Congo et présente une réfraction vert pomme pathognomonique lorsqu'elle est observée à la lumière polarisante (figure 1). Des colorations immunohistochimiques complémentaires ou, mieux encore, la spectrométrie de masse, aident à identifier le type définitif de fibrille amyloïde. Les colorations immunohistochimiques produisent en effet parfois des résultats douteux ou non concluants. En cas d'ATTR confirmée, une analyse de la mutation du gène TTR est toujours indispensable, indépendamment de l'âge de survenue, afin exclure tout type muté d'ATTR et de prévoir éventuellement la mise en place de conseils génétiques1.
Conclusions
L'amylose cardiaque ATTR est beaucoup plus fréquente qu'on ne le pensait dans le passé ; un « indice de suspicion» élevé, souvent basé sur les signaux d'alerte cliniques ou d'imagerie, est nécessaire. De nouveaux outils diagnostiques et une approche par étapes rendent le diagnostic aisé sur le plan pratique. Un diagnostic précoce et correct est essentiel car il existe désormais des traitements permettant d'améliorer considérablement la fonctionnalité et la survie du patient atteint d'ATTR. L'analyse du gène TTR est toujours indispensable et permet au patient et à sa famille de bénéficier de conseils génétiques. Considérer l'ATTR comme une maladie rare et non traitable est une vision désormais obsolète.
Références
- Maurer, M.S., Elliott, P., Comenzo, R., Semigran M., Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation, 2017, 135 (14), 1357-1377.
- Maurer, M.S. , Bokhari, S., Damy, T., Dorbala, S., Drachman, B.M. Fontana, M. et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail, 2019, 12 (9), e006075.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.