ESC-congres 2020
Symposiumverslag
Achtergrond
Cardiale amyloïdose is een infiltratieve cardiomyopathie door progressieve extracellulaire neerslag van onoplosbare amyloïdfibrillen, gevormd uit precursoreiwitten. Het betreft meestal een systeemaandoening waarbij andere orgaansystemen geïnfiltreerd kunnen worden, zoals neuraal, oculair, renaal, gastro-intestinaal en musculoskeletaal, wat zich vertaalt in symptomatische orgaandisfuncties. Twee precursoreiwitten veroorzaken 95 % van de cardiale-amyloïdosegevallen: immuunglobuline-G lichte keten (AL) en meer frequent transthyretine (ATTR), een serumtransporteiwit.1 AL-amyloïd cardiomyopathie is een hematologische maligniteit door monoklonale overproductie en neerslag van immuunglobuline lichte ketens. Transthyretine-amyloïd cardiomyopathie (ATTR-CMP) bestaat uit twee vormen. Wildtype ATTR ontstaat door idiopathische dissociatie van normaal transthyretine-eiwit naar monomeren die amyloïdfibrillen vormen bij voornamelijk Kaukasische mannen (9:1) vanaf 70 jaar (vroeger of later is mogelijk). Het is de meest frequente vorm van cardiale amyloïdose en komt onder andere voor bij 13 % van ouderen met hartfalen en bewaarde ejectiefractie (HFpEF), 16 % bij percutane aortaklepimplantatie (TAVI) en 6 % bij hypertrofische cardiomyopathie.1 Mutanttype (of varianttype) ATTR is het gevolg van abnormaal gevormd transthyretine-eiwit door een genetische mutatie (> 120 mutaties beschreven) in het TTR-gen bij zowel mannen als vrouwen vanaf 40-50 jaar (vroeger en later is mogelijk) en met een zekere geografische en etnische prevalentie. Het onderscheid tussen deze drie vormen van cardiale amyloïdose is essentieel, gezien de sterk verschillende prognose en behandeling. De gemiddelde overleving na een diagnose van symptomatische cardiale amyloïdose bedraagt 6 maanden (AL), 3,5 jaar (wildtype ATTR) en 2-5 jaar (mutanttype ATTR).1 Het onderscheid tussen de vormen berust op een integratie van het klinische profiel, bloedtesten en niet-invasieve cardiale beeldvorming, soms aangevuld door een weefselbiopt. Belangrijk, geen enkel beeldvormend onderzoek kan evenwel definitief het onderscheid maken. Beperkte kennis van de aandoening resulteert in een misdiagnose bij 35 % van de gevallen. Een vroegtijdige diagnose is nochtans prognostisch primordiaal om tijdig chemoen/ of immuuntherapie (AL), dan wel tetrameerstabilisatie en/of TTR-gen silencing (ATTR) aan te bieden.1, 2
Wanneer verdenking op ATTR-CMP?
Infiltratie door amyloïd in de verschillende orgaansystemen leidt tot structurele en functionele veranderingen in de verschillende organen, wat zich vertaalt in een geheel van soms typische of suspecte klinische symptomen/tekenen en afwijkingen op cardiale beeldvorming (tabel 1).
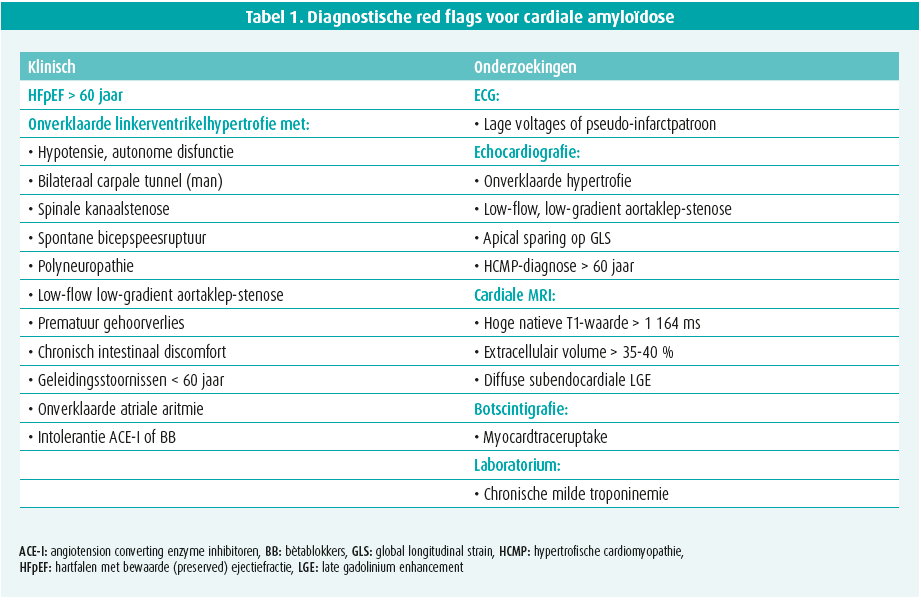
Klinische red flags
Cardiale kliniek omvat vooral HFpEF, zeer vaak als eerste teken en zeker suspect bij mannen boven de 65 jaar. Dit gaat bijna steeds gepaard met een NT-proBNP-stijging en milde troponinemie. Optreden van onverklaarde atriale aritmie en/of conductiestoornissen (pacemakernood), zeker op vroegere leeftijd, zijn tevens suspect. Het ecg is verder aspecifiek maar suspect bij lage voltages en een pseudo-infarctpatroon. Angina, kaakclaudicatio en ischemie door (micro)vasculaire infiltratie, alsook onverklaarde trombo-embolie zijn mogelijk.1
Extracardiale klachten of tekenen van progressieve infiltratie zijn vaak co-existent, dikwijls 5 tot 10 jaar voor de diagnose.1 In deze klinische context is een voorgeschiedenis van carpaaltunnelsyndroom (bilateraal, zeker bij mannen), spinale kanaalstenose, spontane bicepspeesruptuur, springvinger, autonome disfunctie (hypotensie, gastro-intestinaal onverklaard ongemak, gewichtsverlies), gehoorverlies en onverklaarde perifere sensorische en/of motorische polyneuropathie erg suspect. Glaucoom kan ook optreden. Bij mutanttype ATTR staat, afhankelijk van het type mutatie, een klinisch fenotype van neuropathie of cardiomyopathie dan wel een mengvorm op de voorgrond dat zich al kan uiten vanaf de 3de of 4de levensdecade. Specifiekere tekenen bij AL zijn periorbitale purpura, huiduitslag en macroglossie (10 %).
Verdachte cardiale beeldvorming
Echocardiografie toont een milde tot vaak uitgesproken hypertrofie van het linker- (en rechter-)ventrikel met een granulair aspect. Hoewel meestal concentrisch, kan deze hypertrofie licht asymmetrisch zijn, al dan niet met uitstroombaanobstructie. De ejectiefractie van het linkerventrikel blijft meestal behouden tot in een gevorderd stadium van de ziekte, terwijl een matige tot uitgesproken longitudinale disfunctie vaak al in een beginstadium aanwezig is. Deze disfunctie is typisch voor de basale en middelste segmenten maar niet voor de apicale segmenten, wat aanleiding geeft tot het bijna pathognomonische beeld van apical sparing op de 'global longitudinal strain bull's eye plot' (figuur 1). Diastolische disfunctie, tekens van verhoogde vullingsdrukken en biatriale dilatatie zijn frequent. Soms is er milde pericardeffusie en verdikte klepbladen met milde lekkage.1
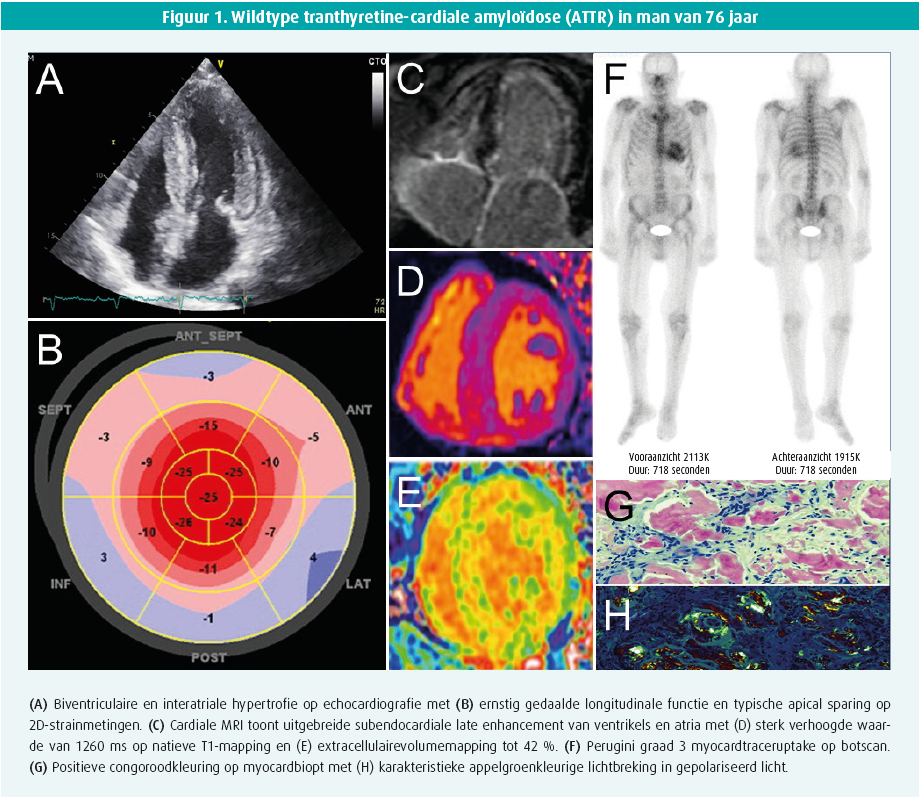
Cardiale MRI biedt naast vaststellen van kamerhypertrofie vooral de meerwaarde van weefselkarakterisatie op basis van 'late gadolinium enhancement' (LGE, maat voor focale fibrose) en extracellulairvolumebepaling (ECV, maat voor diffuse fibrose). LGE is meestal aanwezig, soms als een diffuse subendocardiale schil (pathognomonisch), maar vaker als een eerder vlekkerig of transmuraal patroon. Het ECV is typisch fors verhoogd > 35-40 % (normaal < 25 %) (figuur 1). Daarnaast is een hoge T1-relaxatietijd (zonder contrast), moeilijke nulling over een range van inversietijden, een donkere bloedpool en abnormale/moeilijke suppressie van het vaak diffuus geïnfiltreerd myocard vrij typisch.1, 2
Praktische diagnostiek van ATTR-CMP
Bij enige verdenking van cardiale amyloïdose op basis van klinische red flags, echocardiografie, cardiale MRI en/of biomerkers, volgt stapsgewijze diagnostiek2 (figuur 2).
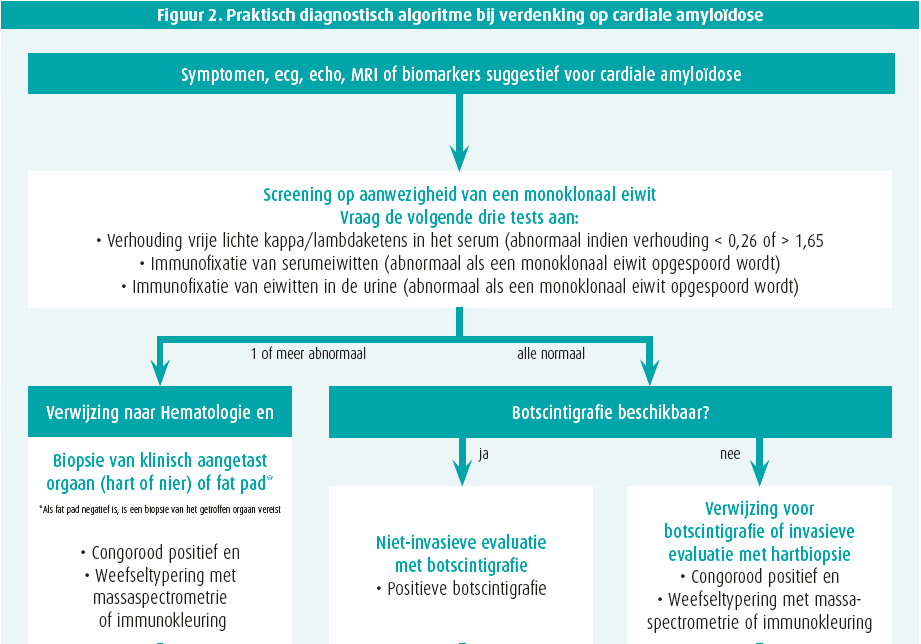
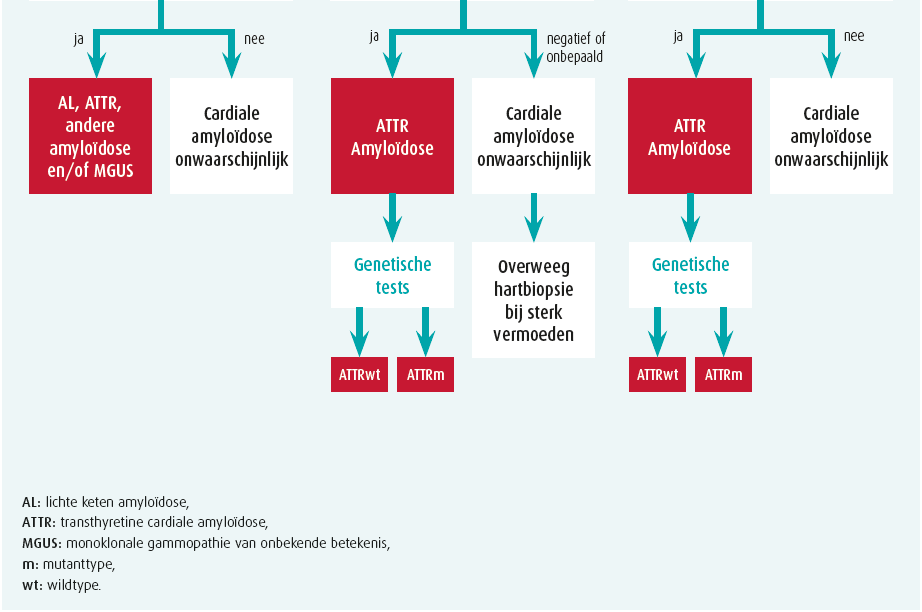
Stap 1: exclusie monoklonaliteit
Er moeten 3 zaken bepaald worden: een bloedstaal en urinestaal telkens met eiwitelektroforese en immuunfixatie (abnormaal als monoklonaal) alsook een vrijelichteketenbepaling op serum (abnormaal als de kappa-lambdaratio < 0,26 of > 1,65 is). Hematologisch advies is aangewezen als een van deze 3 testen abnormaal is, waarbij beenmergbiopsie alsook gerichte (cardiale of renale) orgaanbiopsie nodig kan zijn. Let wel, monoklonale gammopathie van onbekende betekenis (MGUS) komt voor bij 3,2 % 50-plussers en is dus erg vaak co-existent met ATTR.2
Stap 2: botscintigrafie
Technetiumbottracers hebben een (niet geheel verklaarde) affiniteit voor voornamelijk TTR-amyloïd en soms (eerder zwak) voor AL. Een SPECT-botscintigrafie met vraag naar myocardopname alsook tomografische snedes is daarom de volgende stap. Ofwel is er geen myocardtraceruptake (perugini graad 0) ofwel is er uptake die wordt vergeleken met omliggende skelettekening: perugini graad 1 (minder dan skelet), 2 (gelijk aan skelet) of 3 (meer dan skelet). In afwezigheid van monoklonaliteit, heeft graad 2 of meer uptake 100 % predictieve waarde voor ATTR waardoor een eventuele noodzaak aan myocardbiopsie vervalt1, 2 (figuur 1). Graad 0 of 1 uptake is evenwel mogelijk bij zowel AL of (vroeg stadium) ATTR en vergt orgaanbiopsie. Verschillende PET-scantracers zijn inmiddels ook veelbelovend voor amyloïdosediagnostiek, maar worden nu vooral toegepast voor onderzoeksdoeleinden.
Stap 3: bevestiging cardiale amyloïdose
ATTR staat vast als de myocardtraceruptake zonder dat monoklonaliteit graad 2 of meer is. Andere situaties vergen biopsie. Een vetbiopt is makkelijk, maar heeft beperkte sensitiviteit (AL 84 %, mutanttype ATTR 45 % en wildtype ATTR slechts 15 %).1 Een gerichte biopsie van een aangedaan orgaan is daarom veel sensitiever (renaal of cardiaal). De cardiale amyloïd-infiltratie is niet steeds homogeen en LGE cardiale MRI kan een leidraad vormen om sampling-error (valsnegatief resultaat) te vermijden. Amyloïd kleurt aan na congoroodkleuring en vertoont een pathognomonische appelgroenkleurige lichtbreking onder gepolariseerd licht (figuur 1). Aanvullende immuunhistochemische kleuringen, of beter nog, massaspectrometrie helpen het finale type van amyloïdfibril te identificeren. Immuunhistochemische kleuringen geven immers soms dubieuze of inconclusieve resultaten. Bij bevestigde ATTR is steeds een TTR-genmutatieanalyse noodzakelijk, onafhankelijk van de leeftijd van presentatie, om een mutanttype ATTR uit te sluiten en eventuele genetische counseling te voorzien.1
Conclusies
ATTR cardiale amyloïdose is veel frequenter dan vroeger werd aangenomen en een hoge 'index of suspicion' is noodzakelijk, vaak op basis van klinische of beeldvormende red flags. Nieuwe diagnostische middelen en een stapsgewijze aanpak maken de diagnose praktisch makkelijk. Vroegtijdige en correcte diagnostiek is essentieel, aangezien er nu therapieën beschikbaar zijn die de functionaliteit en overleving van de ATTR-patiënt aanzienlijk kunnen verbeteren. TTR-genanalyse is steeds obligaat en laat genetische counseling toe van de patiënt en de familie. De conceptie van ATTR als een zeldzame, onbehandelbare aandoening is obsoleet.
Referenties
- Maurer, M.S., Elliott, P., Comenzo, R., Semigran M., Rapezzi, C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation, 2017, 135 (14), 1357-1377.
- Maurer, M.S. , Bokhari, S., Damy, T., Dorbala, S., Drachman, B.M. Fontana, M. et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail, 2019, 12 (9), e006075.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.