Congrès de l'ESC 2020
Compte rendu du symposium
Cette session était présidée par le Pr Pablo Garcia-Pavia (Madrid, Espagne) et le Pr Thibaud Damy (Créteil, France).
Lors de cette session, on a traité plus en détail les notions et les possibilités thérapeutiques les plus récentes pour les patients souffrant d'une cardiomyopathie amyloïde liée à la transthyrétine (ATTR-CM).
Dans la première partie de la session, le Pr Pablo Garcia-Pavia a discuté des options de traitement médical actuelles. L'ATTR-CM est une affection progressive et mortelle, et les patients diagnostiqués aujourd'hui ne représentent que la partie émergée de l'iceberg. Les études indiquent de plus en plus que l'ATTR-CM est plus fréquente qu'on ne le pense. Ainsi, dans une étude, la prévalence des dépôts d'amyloïde dans le coeur atteignait même 10 % chez les sujets de plus de 65 ans souffrant d'insuffisance cardiaque à fraction d'éjection préservée, ayant une hypertrophie ventriculaire gauche de 15 mm ou plus1. Des chiffres similaires ont également été trouvés chez les patients ayant bénéficié d'une implantation de valve aortique transcathéter (TAVI).
Jusqu'il y a peu, on disposait principalement de traitements médicaux de soutien pour cette affection, qui ne traitent que les conséquences mais n'agissent pas sur la cause sous-jacente. La pierre angulaire du traitement de l'insuffisance cardiaque consiste toujours à éviter la congestion ou l'accumulation de liquide grâce à un régime pauvre en sel avec restriction hydrique et pesée quotidienne pour optimiser les doses de diurétiques. à ce jour, il n'existe également pas de preuves que le traitement classique de l'HFrEF (insuffisance cardiaque à fraction d'éjection réduite) e.a. par inhibiteurs de l'ECA, sartans, bêtabloquants, antagonistes du récepteur des minéralocorticoïdes ou ivabradine ait un quelconque effet clinique ou pronostique. Au contraire, des données montrent que ces médicaments peuvent aggraver l'évolution de l'ATTR-CM. En effet, l'expérience nous a appris que les inhibiteurs de l'ECA ou les sartans sont mal tolérés en raison de l'hypotension. Il en va certainement de même pour les bêtabloquants, après l'arrêt desquels les patients s'améliorent souvent cliniquement, car le débit cardiaque lors d'une telle cardiomyopathie restrictive dépend davantage de la fréquence cardiaque. Un autre aspect important en cas d'ATTR-CM est la prévention et le traitement des arythmies auriculaires. Ainsi, une étude démontre que le maintien du rythme sinusal après un épisode de fibrillation auriculaire pourrait améliorer le pronostic de ces patients. Cependant, la cardioversion électrique n'est pas dénuée de risques, et jusqu'à 20 % des patients peuvent même présenter des complications telles que des troubles du rythme ventriculaire, un bloc AV transitoire et un AVC ischémique. Le succès de la cardioversion dépend surtout du stade de l'ATTR-CM dans lequel se trouve le patient. Plus la maladie est déjà marquée et avancée (selon l'échelle de Gillmore2), moins il y a de chances de maintien du rythme sinusal. En outre, on ne sait pas exactement à partir de quand les patients doivent être anticoagulés, étant donné qu'une autre étude basée sur l'IRM a révélé un nombre important de thrombi intracardiaques, même en rythme sinusal (comme en cas de sténose valvulaire mitrale). Par ailleurs, on ignore actuellement quels anticoagulants (warfarine vs DOAC) sont les plus efficaces à cet égard.
Le Pr Thibaud Damy a ensuite abordé le rôle du tafamidis pour le traitement de l'ATTR-CM. Le tafamidis inhibe les dépôts d'amyloïde dans les organes en se liant sélectivement au site de liaison à la thyroxine de la protéine transthyrétine (TTR) plasmatique, ce qui empêche sa dissociation en monomères ; en effet, ce sont ces monomères qui forment la base des dépôts d'amyloïde. Sur la base de l'étude ATTR-ACT, le tafamidis à la dose de 61 mg une fois par jour a été approuvé par l'EMA pour le traitement de l'amyloïdose cardiaque liée à la transthyrétine de type sauvage (WT) et héréditaire chez les adultes3.
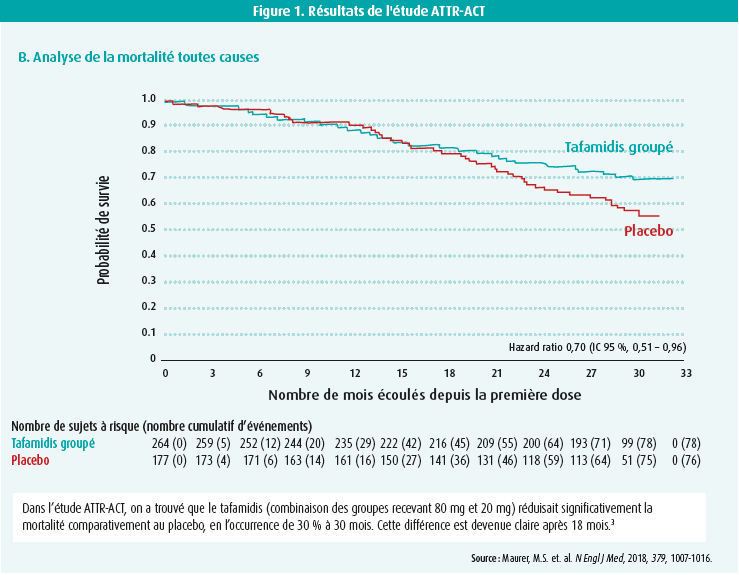
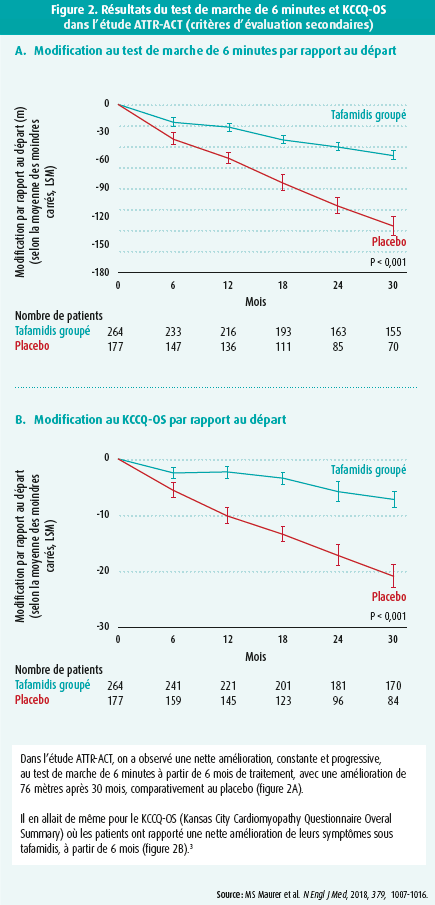
En outre, le tafamidis 61 mg est le bioéquivalent approuvé du tafamidis méglumine 80 mg (20 mg 4 fois par jour) utilisé dans l'étude ATTR-ACT. Les résultats de cette étude avaient été présentés lors du congrès de la ESC à Munich en 2018 et publiés simultanément dans le New England Journal of Medicine. Dans cette étude multicentrique, 441 patients atteints d'ATTR-CM WT (n = 335) ou héréditaire (n = 106) ont été évalués de manière randomisée, en double aveugle et versus placebo. Ces patients ont été randomisés en trois groupes : tafamidis méglumine 80 mg/jour, 20 mg/jour et placebo. à cet égard, on a découvert que, dans l'ensemble du groupe tafamidis, la combinaison de la mortalité et des hospitalisations liées à des problèmes cardiovasculaires diminuait significativement par rapport au placebo après 30 mois. Malgré cela, l'ATTR-CM reste une affection dont le pronostic est limité, 71 % des patients étant encore en vie au bout de 30 mois dans le groupe tafamidis, contre 57 % dans le groupe placebo. La diminution relative de la mortalité atteignait 30 % par rapport au placebo, avec un NNT (number needed to treat) de 7,5 (figure 1). Le tafamidis est donc le premier traitement qui a pu démontrer une réduction de la mortalité en cas d'ATTR-CM. En outre, on a également découvert que le tafamidis entraînait une réduction de 32 % du risque relatif d'hospitalisations pour problèmes cardiovasculaires après 30 mois, par rapport au placebo. Les paramètres subjectifs et cliniques s'amélioraient également. Ainsi, on a observé une nette amélioration, constante et progressive, au test de marche de 6 minutes à partir de 6 mois de traitement, avec une amélioration de 76 mètres après 30 mois, comparativement au placebo. Il en allait de même pour le KCCQ-OS (Kansas City Cardiomyopathy Questionnaire Overal Summary) où les patients ont rapporté une nette amélioration de leurs symptômes sous tafamidis, à partir de 6 mois (figures 2A et 2B).
Après 30 mois, les patients ont eu la possibilité de participer à l'étude d'extension ATTR-ACT dans laquelle le groupe placebo a été randomisé selon un rapport 2:1 vers 80/20 mg de tafamidis. à partir du 1er juillet 2018, tous les patients sont passés à la nouvelle formulation de 61 mg une fois par jour (bioéquivalent du tafamidis méglumine 80 mg). Les résultats de cette étude ont été présentés par le Pr Damy au Congrès européen sur l'insuffisance cardiaque en juin de cette année. Ces données montrent que le tafamidis à la dose de 80 mg offrait une meilleure survie à long terme, comparativement à la dose de 20 mg, après 51 mois, avec une réduction du risque de 30 %, qui est par conséquent la posologie de choix.
Le profil de sécurité du tafamidis est également apparu favorable, avec des notifications d'effets indésirables globalement similaires par rapport au placebo. Les flatulences et de légères perturbations des tests hépatiques étaient un peu plus fréquentes sous tafamidis. Des données récentes indiquent également que le tafamidis à la dose de 61 mg abaisse la thyroxine sérique totale, sans effet sur la T4 libre ou la TSH.
La présentation de ces données a été suivie d'une session de questions-réponses en ligne en direct avec les deux modérateurs. Quelques questions intéressantes et pratiques liées au diagnostic et au traitement de l'ATTR-CM ont été posées.
En pratique, il est recommandé de débuter le traitement par tafamidis dès qu'une atteinte cardiaque peut être prouvée. Plus le traitement peut être débuté tôt, mieux c'est (NYHA I et II), car l'étude ATTR-ACT montre que c'est ce groupe qui en tire le plus de bénéfices. Toutefois, il y a également des éléments (basés sur l'étude d'extension ATTR-ACT) qui indiquent qu'on peut aussi attendre un effet bénéfique en classe NYHA III4. Cependant, sur ce plan, il ne faut pas espérer de réversibilité sur le coeur ou d'autres organes, ce qui explique que le tafamidis n'est pas indiqué en classe NYHA IV (c'était un critère d'exclusion de l'étude ATTR-ACT). Cela peut également expliquer pourquoi l'effet sur la mortalité n'a commencé à être visible qu'après environ 18 mois (figure 1).
En pratique, pour le diagnostic de l'ATTR-CM, une scintigraphie osseuse positive avec une fixation cardiaque claire (grade 2 ou 3) et l'exclusion d'une amyloïdose AL sous-jacente chez les patients souffrant d'hypertrophie cardiaque sont souvent suffisantes5. Cependant, il existe d'autres causes rares pouvant entraîner une fixation cardiaque faussement positive, comme une toxicité de l'hydroxychloroquine, qui doivent également être exclues. Toutefois, une biopsie cardiaque peut être nécessaire si l'imagerie est douteuse (p. ex. si elle est (faussement) négative en cas de forte suspicion clinique). Quel que soit l'âge du patient, un test génétique est toujours recommandé pour exclure une forme héréditaire d'ATTR-CM, dans le cadre d'un conseil génétique pour les membres de la famille.
Pour les porteurs génétiques asymptomatiques d'une anomalie de la TTR, on recommande d'attendre jusqu'à ce que les dépôts d'amyloïde cardiaques soient démontrés. Dans le rare cas d'une neuropathie combinée à une cardiomyopathie amyloïde familiale liée à la transthyrétine, le Pr Damy préconise également le tafamidis à la dose de 61 mg au lieu de 20 mg.
Note de l'auteur
Au moment de la rédaction de cet article, le tafamidis n'est pas encore remboursé en Belgique pour le traitement de l'ATTR-CM, mais ce remboursement est prévu dans le courant de l'année prochaine. Cependant, en attendant, la firme Pfizer propose un programme médical d'urgence depuis le 1er août 2020.
Referenties
- Damy, T., Costes, B., Hagège, A.A., Donal, E., Eicher, J.C., Slama, M., et al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J, 2016, 37>, 1826-1834.
- Gillmore, J.D., Damy, T., Fontana, M., Hutchinson, M., Lachmann, H.J., Martinez- Naharro, A., et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J, 2018, 39, 2799-2806.
- Maurer, M.S.,, Schwartz J.H., Gundapaneni, B., Elliott, P.M., Merlini, G., Waddington-Cruz, M., et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med, 2018, NEJMoa1805689.
- Rapezzi, C,. ESC 2020 e-poster: Benefits of tafamidis in patients with advanced transthyretin amyloid cardiomyopathy.
- Hanna, M., Ruberg, F.L., Maurer, M.S., Dispenzieri, A., Dorbala, S., Falk, R.H., et al. Cardiac Scintigraphy With Technetium-99m- Labeled Bone-Seeking Tracers for Suspected Amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol, 2020, 75, 2851-2862.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.