Compte rendu d'une session de la BSC - session 4 - BWGBRC
Sandrine Horman et Michel De Pauw ont modéré la session du BWGBRC (Belgian Working Group of Basic Research in Cardiology) lors du congrès 2021 de la Société Belge de Cardiologie. E. Dale Abel (University of Iowa City, USA) a ouvert la session avec une présentation axée sur les altérations métaboliques qui caractérisent le coeur diabétique. Christian Schulze (University Hospital of Jena, Allemagne) a ensuite discuté de la lipotoxicité associée à l'insuffisance cardiaque. Enfin, Walter Paulus (University Medical Center, Amsterdam, Pays-Bas) a présenté des évidences expérimentales soutenant le lien existant entre l'inflammation, les altérations microvasculaires et la dysfonction diastolique des patients souffrant d'insuffisance cardiaque avec fraction d'éjection préservée.
Le coeur diabétique
Une des caractéristiques majeures du métabolisme cardiaque est sa flexibilité. En effet, le coeur est capable d'utiliser de nombreuses sources d'énergie dont les principales sont les acides gras, le glucose, le lactate, les corps cétoniques et certains acides aminés. Le choix de la source d'énergie dépend de la disponibilité des substrats, des besoins énergétiques et de la situation hormonale. A jeun, le coeur produit son énergie en oxydant les acides gras, alors que l'oxydation du glucose fournit l'énergie en situation post-prandiale. Cette flexibilité métabolique est essentielle au maintien de l'intégrité de la cellule cardiaque et de la machinerie contractile. Elle est cependant compromise dans le coeur diabétique, entraînant l'apparition d'une pathologie appelée cardiomyopathie diabétique, indépendante de la maladie coronarienne et de l'hypertension artérielle.
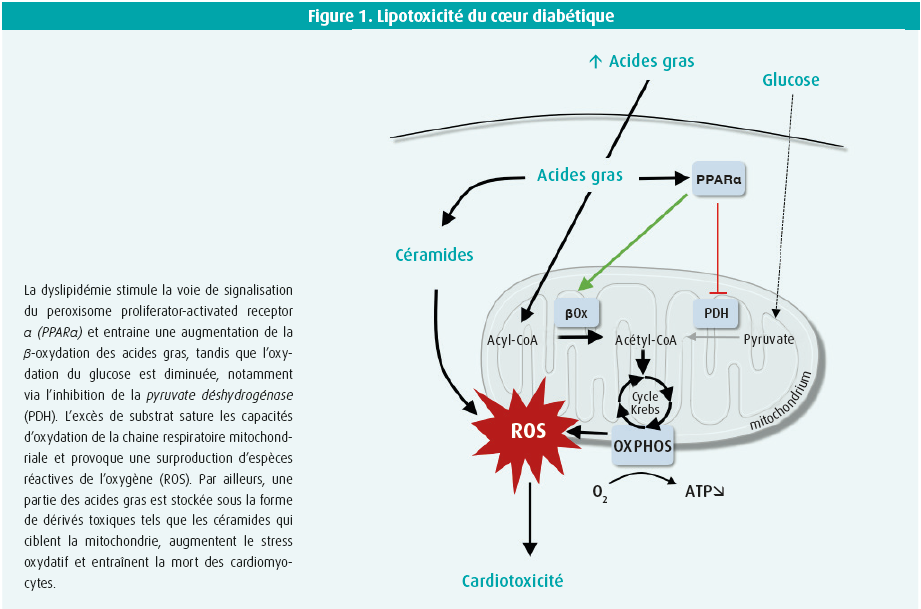
Depuis de nombreuses années, les recherches de E. Dale Abel ont mis en évidence une multiplicité de facteurs physiopathologiques liant les changements d'environnement du myocarde induits par le diabète et la dysfonction cardiaque1. Parmi eux, la dyslipidémie, l'hyperglycémie et la résistance à l'insuline vont contribuer à l'apparition d'une inflexibilité métabolique. En effet, malgré la présence d'une hyperglycémie, le coeur diabétique utilise majoritairement les acides gras circulants pour produire la totalité de son énergie, et ce, quelles que soient les conditions hormonales ou nutritionnelles. L'augmentation incontrôlée de l'oxydation des acides gras entraîne alors une production massive d'équivalents réducteurs (NADH et FADH2) qui dépassent les capacités d'oxydation de la chaine respiratoire et entraînent une surproduction d'espèces réactives de l'oxygène. Ce stress oxydatif aggrave la dysfonction mitochondriale, ce qui réduit encore l'apport énergétique nécessaire au bon fonctionnement de la machinerie contractile. Plusieurs mécanismes participent à l'apparition de cette inflexibilité métabolique parmi lesquels l'insulino-résistance et l'activation par les lipides de la voie de signalisation du peroxisome proliferator-activated receptor α (PPAR α) (figure 1). La cardiomyopathie diabétique est également caractérisée par l'accumulation d'acides gras au sein du cardiomyocyte. On les retrouve sous la forme de triglycérides mais aussi de dérivés lipidiques toxiques tels que les céramides qui ciblent la mitochondrie, augmentent le stress oxydatif et entraînent la mort des cardiomyocytes par apoptose (figure 1).
E. Dale Abel a aussi mis en exergue les effets néfastes de l'hyperglycémie, et notamment l'augmentation de l'O-GlcNacylation des protéines, une modification post-traductionnelle affectant, entre autres, la réabsorption calcique au niveau du réticulum sarcoplasmique, et pouvant mener à une perte de contractilité via l'altération de l'activation de la protéine kinase IIδ dépendante du calcium et de la calmoduline (CaMKIIδ), un régulateur majeur de la fonction cardiomyocytaire. Par ailleurs, l'excès de glucose entraîne un changement d'expression génique induisant également un profond remodelage du métabolisme mitochondrial cardiaque en faveur de l'oxydation des acides gras2.
L'ensemble de ces altérations aboutit à une dysfonction, principalement diastolique, entraînant une augmentation des risques d'insuffisance cardiaque.
Lipotoxicité et insuffisance cardiaque
La perte de flexibilité métabolique du coeur est également une des caractéristiques de l'insuffisance cardiaque. On observe ainsi dans ces conditions une diminution de l'oxydation des acides gras favorisant l'accumulation dans le cardiomyocyte d'intermédiaires lipidiques toxiques. Au cours de sa présentation, Christian Schulze a exposé plusieurs résultats clés obtenus dans le cadre d'études menées par son équipe de recherche sur la lipotoxicité associée à l'insuffisance cardiaque3. Il a notamment démontré que cette pathologie s'accompagne d'une augmentation des niveaux circulants et cardiaques d'acylcarnitines à très longues chaînes, ce qui corrèle d'ailleurs avec la sévérité de la maladie4. Les céramides à très longues chaînes sont un autre exemple d'intermédiaires lipidiques augmentés dans le plasma et le coeur des patients. Ils sont constitués d'une molécule de sphingosine couplée à un acide gras et sont synthétisés via trois grandes voies. L'une de ces voies (synthèse de novo) implique la condensation d'un palmitoyl- CoA et d'une sérine par la sérine palmitoyltransférase (SPTLC2), enzyme limitante de la voie dont l'expression est augmentée en cas d'insuffisance cardiaque. L'inhibition pharmacologique de cette enzyme par la myriocine, de même que la délétion du gène SPTLC2 dans un modèle murin, entraînent une baisse des niveaux de céramides et une prévention de l'insuffisance cardiaque post-infarctus5. A l'inverse, la surexpression de la céramide synthase (CerS2) catalysant la formation de céramides à partir de sphingosine (voie de sauvetage) est associée à une accumulation de céramides ayant pour conséquence l'activation de voies pro-apoptotiques, des troubles du métabolisme oxydatif ou encore des événements de mitophagie6. Ces travaux ont permis de mettre en évidence des mécanismes moléculaires qui pourraient être la cible de nouvelles stratégies thérapeutiques visant à manipuler le métabolisme afin de limiter la progression de l'insuffisance cardiaque.
Rôle central de l'endothélium microvasculaire dans la physiopathologie de l'HFpEF
L'insuffisance cardiaque est majoritairement décrite sous deux formes : (1) une dysfonction systolique accompagnée d'une dilatation du myocarde qui entraînent une diminution de la fraction d'éjection (HFrEF) ; (2) une dysfonction diastolique, faisant suite à une hypertrophie concentrique du muscle cardiaque et une élévation de la pression de remplissage sans perturbation de la fraction d'éjection (HFpEF)7. Alors que l'HFrEF se développe principalement lors de circonstances pathologiques qui altèrent l'intégrité du cardiomyocyte (comme par exemple un infarctus du myocarde), il reste difficile de pointer une cause bien déterminée de l'HFpEF. Il est reconnu que la prévalence de comorbidités métaboliques telles que le diabète est très élevée chez les patients HFpEF. Cette observation a donné naissance au paradigme selon lequel ces comorbidités métaboliques peuvent entraîner une inflammation chronique de bas grade à l'origine d'une activation de l'endothélium des microvaisseaux coronaires et d'un stress oxydatif qui contribuent, via le découplage de la oxyde nitrique synthase (NOS), à l'augmentation de la taille et de la rigidité passive des cardiomyocytes8,9.
La contribution centrale de l'endothélium à la rigidité myocardique typique de l'HFpEF est notamment soutenue par les données récentes de l'étude MESA qui corrèlent fortement la dysfonction diastolique au syndrome métabolique des patients HFpEF diabétiques, et ce, indépendamment de la fibrose cardiaque, récapitulant ainsi les données obtenues par le groupe de Walter Paulus en 200810. Plus récemment et en accord avec ce paradigme, Frisk et al. ont examiné les caractéristiques des tubules-T chez les patients HFpEF diabétiques et mis en évidence une augmentation du temps de réabsorption du Ca2+ par les tubules-T, par comparaison aux patients non diabétiques11. Enfin, une étude récente révèle que les patients HFpEF n'évoluent pas ou peu (< 2 % sur une période de 11 ans) vers un phénotype HFrEF, indiquant clairement que la dysfonction diastolique n'est pas le précurseur d'une dilatation ventriculaire et d'une diminution de la fonction systolique12.
Références
- Ritchie, R.H., Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ Res, 2020, 126 (11), 1501-1525.
- Wende, A.R., Schell, J.C., Ha, C.M., Pepin, M.E., Khalimonchuk, O., Schwertz, H. et al. Maintaining Myocardial Glucose Utilization in Diabetic Cardiomyopathy Accelerates Mitochondrial Dysfunction. Diabetes, 2020, 69 (10), 2094-2111.
- Kretzschmar, T., Wu, J.M.F., Schulze, P.C. Mitochondrial Homeostasis Mediates Lipotoxicity in the Failing Myocardium. Int J Mol Sci, 2021, 22 (3), 1498.
- Goldenberg, J.R., Carley, A.N., Ji, R., Zhang, X., Fasano, M., Schulze, P.C. et al. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation, 2019, 139 (24), 2765-2777.
- Ji, R., Akashi, H., Drosatos, K., Liao, X., Jiang, H., Kennel, P.J. et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight, 2017, 2 (14).
- Law, B.A., Liao, X., Moore, K.S., Southard, A., Roddy, P., Ji, R. et al. Lipotoxic verylong- chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J, 2018, 32 (3), 1403- 1416.
- Pieske, B., Tschope, C., de Boer, R.A., Fraser, A.G., Anker, S.D., Donal, E. et al. How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J, 2019, 40 (40), 3297-3317.
- Paulus, W.J., Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol, 2013, 62 (4), 263-271.
- van Heerebeek, L., Hamdani, N., Handoko, M.L., Falcao-Pires, I., Musters, R.J., Kupreishvili, K., et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation, 2008, 117 (1), 43-51.
- Ladeiras-Lopes, R., Moreira, H.T., Bettencourt, N., Fontes-Carvalho, R., Sampaio, F., Ambale- Venkatesh, B. et al. Metabolic Syndrome Is Associated With Impaired Diastolic Function Independently of MRI-Derived Myocardial Extracellular Volume: The MESA Study. Diabetes, 2018, 67 (5), 1007-1012.
- Frisk, M. Le, C., Shen, X., Roe, A.T., Hou, Y., Manfra, O. et al. Etiology-Dependent Impairment of Diastolic Cardiomyocyte Calcium Homeostasis in Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol, 2021, 77 (4), 405-419.
- Lupon, J., Gavidia-Bovadilla, G., Ferrer, E., de Antonio, M., Perera-Lluna, A., Lopez-Ayerbe, J. et al. Heart Failure With Preserved Ejection Fraction Infrequently Evolves Toward a Reduced Phenotype in Long-Term Survivors. , 2019, 12 (3), e005652.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.