Introduction
Le décès subit et souvent inattendu de membres de la famille ou de connaissances est un problème connu qui ne laisse personne indifférent. Lorsque le décès survient dans l'heure qui suit l'apparition des symptômes, et qu'il est la conséquence d'une affection cardiaque, on parle de mort cardiaque subite. Nous avons tous entendu les médias relater des cas de mort subite touchant des personnes souvent jeunes, apparemment en bonne santé, qui seraient décédées d'une « insuffisance cardiaque » ou d'une « rupture de l'aorte ». La cause exacte reste souvent indéterminée et même après l'autopsie, il est fréquent qu'aucune étiologie définitive ne puisse être mise en évidence, ce qui donne assez souvent lieu au diagnostic final aspécifique de « arrêt cardiaque ».
Avec une incidence annuelle d'un ou de deux décès par mille annéespersonnes, la mort cardiaque subite a des répercussions sociales majeures. En Belgique, ce chiffre représente environ 5 à 10 % de l'ensemble des causes de décès naturelles, soit 10 000 décès par an.
Notre population n'est pas systématiquement formée pour prodiguer des soins de premiers secours appropriés ; or, l'absence ou l'arrivée tardive d'intervenants capables de pratiquer la réanimation cardiopulmonaire (RCP) réduit sérieusement les chances de survie : après une réanimation, cellesci ne dépassent pas 4 à 10 % et, en cas de rétablissement efficace de la circulation spontanée, les chances de rétablissement sans lésions neurologiques résiduelles demeurent très limitées1, 2. Il s'agit souvent de personnes actives jeunes, ce qui se traduit par un énorme impact socioéconomique et sociétal chaque année.
Malgré notre réseau de soins d'urgence bien développé, les connaissances médicales les plus récentes et un accès aisé à la prise en charge et au soutien cardiaques de pointe, la mort cardiaque subite paraît mal maîtrisée en tant qu'entité clinique. En effet, notre diagnostic cardiaque n'est pas en mesure de rendre compte de façon satisfaisante des manifestations cliniques ; nous avons donc pris pour habitude de travailler avec des diagnostics tels que « cardiomyopathie dilatée idiopathique » ou « FV idiopathique ». Or, derrière ces ensembles de manifestations idiopathiques se cachent peut-être des affections uniques, nécessitant une prise en charge plus spécifique qui permettrait, moyennant l'adoption de mesures appropriées, d'améliorer la prévention de la mort cardiaque subite. Par ailleurs, notre système de stratification de risques basé sur des indicateurs cliniques classiques ne semble pas toujours en mesure d'estimer de façon fiable un risque fortement accru de mort cardiaque subite. Cette situation a conduit à élaborer une stratégie de prévention axée sur l'implantation d'un défibrillateur cardiaque implantable (ICD), suite à laquelle on n'observe souvent plus aucun cas d'arythmie, alors que les patients non porteurs d'un tel ICD présentent encore des événements malins malgré un suivi cardiaque rigoureux, car ils ont été considérés comme à « faible » risque. Il faut réaliser qu'avec la stratification de risques actuelle, lorsque nous utilisons le seuil de risque de mort cardiaque subite de 6 % sur 5 ans pour une implantation d'ICD, nous protégeons seulement la moitié des patients atteints de cardiomyopathie hypertrophique à haut risque3.
En Belgique en outre, il est très rare que des autopsies soient réalisées après une mort cardiaque subite. On part du principe qu'en cas de décès naturel, rechercher la cause du décès ne présente aucun intérêt. Lorsqu'une autopsie est malgré tout pratiquée, la cause du décès demeure souvent inconnue en raison de l'absence d'anomalies cardiaques structurelles manifestes, ce qui aboutit à un diagnostic de « mort subite inexpliquée » (SUDS : Sudden Unexplained Death Syndrome). Lorsqu'un décès « suspect » donne lieu à une autopsie judiciaire, l'autopsie vise exclusivement à exclure une cause criminelle du décès et non à détecter une cause de décès cliniquement pertinente. Tant pour les autopsies civiles que judiciaires, on ne prélève pas d'échantillon d'ADN de manière systématique et la possibilité de mener un examen cardiogénétique post-hoc chez le proband est irrévocablement perdue. Enfin, lorsque le proband est décédé, les membres de la famille sont rarement orientés vers des spécialistes pour passer des examens cardiogénétiques plus poussés.
En un mot, on ne saisit pas les occasions précieuses qui se présentent, ce qui explique le long chemin à parcourir pour améliorer la prise en charge et la prévention de la mort cardiaque subite. Il a cependant été démontré que le dépistage et la prise en charge tertiaires de la mort cardiaque subite peuvent s'avérer rentables pour les survivants et/ou leur famille4. Cet article présente un aperçu des connaissances actuelles en matière de mort subite, des causes connues (l'accent étant mis sur les aspects génétiques), ainsi que des opportunités et des lacunes de notre système de santé, et ce dans le but de mettre en place une meilleure prévention de la mort subite.
Étiologie
Dans 9 cas sur 10, la mort cardiaque subite est la conséquence directe d'arythmies cardiaques malignes tachycardie ventriculaire (TV) et/ou fibrillation ventriculaire (FV), qui représentent la 'final common pathway' (voie finale commune) de pathologies cardiaques pré-existantes. Il ne s'agit que rarement d'une défaillance aiguë de la fonction pompe du coeur ou d'anomalies structurelles catastrophiques, telles qu'une rupture ventriculaire.
L'événement de décès n'est souvent précédé d'aucun symptôme, ou bien les symptômes ne sont pas identifiés comme un signal d'alarme cardiaque, de sorte que les patients n'ont généralement subi aucun examen cardiaque auparavant.
Dès lors, dans 30 % des cas, la mort cardiaque subite constitue le premier et unique symptôme de la cardiopathie sousjacente. Lorsque le patient survit à l'épisode, les contrôles cardiaques cliniques ne détectent pas toujours des anomalies cardiaques. De même, lorsque le patient est décédé, il est fréquent que l'autopsie n'identifie aucune cause claire de décès.
De manière générale, la cause sousjacente de la mort cardiaque subite est, dans environ 60 à 80 % des cas, imputable à une cardiopathie ischémique. Lorsque nous ventilons ces causes en fonction de l'âge, nous remarquons qu'au sein du groupe d'âge de > 45 ans, l'immense majorité des cas de mort cardiaque subite résulte d'un syndrome coronarien aigu (SCA).
Lorsque la mort cardiaque subite touche des personnes de moins de 45 ans, une cause héréditaire sousjacente peut être mise en évidence dans 80 % des cas5. Il s'agit là d'un élément important dans la mesure où l'événement de mort cardiaque subite chez le patient index (patient de référence) doit dès lors être considéré comme un indicateur de la présence d'une maladie cardiaque familiale ; cela signifie qu'il pourrait y avoir d'autres patients à haut risque de la même famille susceptibles de bénéficier de mesures préventives. Il s'agit donc là d'une opportunité importante qui, si elle est saisie, permettrait d'améliorer la prévention de la mort cardiaque subite. Il est donc également crucial d'identifier la cause de la mort cardiaque subite chez le patient index ou le proband, afin de pouvoir adopter les mesures de prévention appropriées pour le proband et de procéder à une stratification ciblée des risques chez les membres de la famille concernés. Dans la pratique, une cause est identifiée dans la moitié des cas seulement ; le décès est donc qualifié de mort subite inexpliquée ou SUDS, et aucun examen clinique familial ciblé n'est possible. Par conséquent, des opportunités importantes de prévention pour les membres de la famille risquent d'être perdues du fait que l'identification d'autres personnes à haut risque reste inconnue.
Dans ce groupe d'âge, nous pouvons subdiviser l'étiologie principale en plusieurs causes distinctes : le premier groupe concerne les cardiopathies structurelles héréditaires, tandis que le deuxième groupe inclut les maladies héréditaires liées à des cardiopathies électriques primitives, pour lesquelles les techniques diagnostiques actuelles ne détectent généralement aucune cardiopathie structurelle évidente (figure 1). Le troisième groupe à l'origine de la mort cardiaque subite, moins fréquemment observé, inclut les anévrismes de l'aorte thoracique et les dissections aortiques (AATDA).
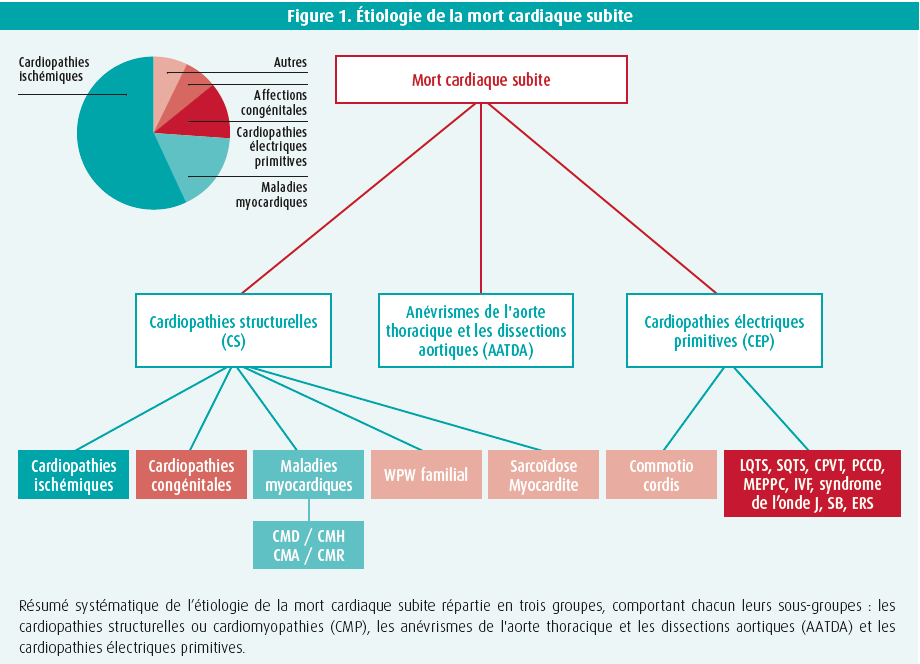
Les maladies myocardiques héréditaires
Les cardiomyopathies incluent un groupe hétérogène de pathologies caractérisées par un remodelage structurel qui compromet progressivement la stabilité électrique du coeur. La cardiopathie structurelle primitive provoque ainsi un risque accru d'arythmie maligne et de mort cardiaque subite. Les cardiomyopathies héréditaires associées à la mort cardiaque subite peuvent se subdiviser en plusieurs groupes : la cardiomyopathie hypertrophique (CMH), la cardiomyopathie dilatée (CMD), la cardiomyopathie arythmogène (CMA) et la cardiomyopathie restrictive (CMR).
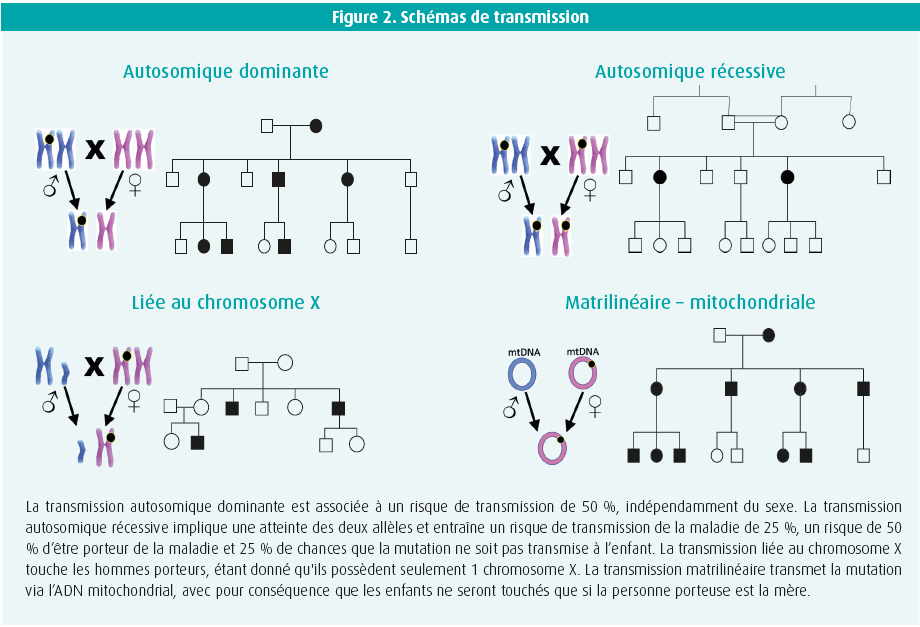
La plupart de ces maladies se transmettent selon le mode autosomique dominant (AD) (1 allèle touché, risque de transmission de 50 %) ; cependant, de rares maladies autosomiques récessives (AR) (2 allèles touchés), ainsi de rares formes liées au chromosome X et formes mitochondriales ont été décrites (figure 2). La présence d'un variant génétique ne signifie pas automatiquement que les signes ou symptômes de la maladie apparaîtront. On parle alors de « pénétrance incomplète ». Les mécanismes sous-jacents ne sont pas bien élucidés.
La cardiomyopathie hypertrophique (CMH)
La CMH est définie comme un épaississement excessif et asymétrique du ventricule gauche en l'absence d'une charge anormale. Avec une prévalence de 1 personne sur 500, cette cardiomyopathie est l'une des cardiomyopathies héréditaires les plus fréquentes, qui se transmet selon le mode autosomique dominant. De ce fait, tout parent du premier degré d'un patient atteint de CMH a 50 % de risque d'être porteur de la même prédisposition génétique de développer le phénotype de CMH.
Cette maladie varie en termes d'âge d'expression et de manifestations cliniques. Même au sein de familles présentant une mutation avérée, il est difficile de prédire si et dans quelle mesure le phénotype sera exprimé. De même, il n'est pas possible de prédire chez des patients individuels si le phénotype en question évoluera vers une insuffisance cardiaque systolique terminale, une situation qui se produit dans environ 5 à 10 % des cas de CMH. Une mort cardiaque subite a été décrite dans 0,4 à 1 % des cas de CMH, mais un risque nettement plus élevé a été observé dans certaines sous-populations. La stratification des risques cliniques actuelle ne permet pas d'estimer de façon précise le risque individuel.
La CMH est causée par des anomalies génétiques au niveau des sarcomères (protéines) (MYBPC3, MYH7, TNNT2, TNNI3 et MYL2), des protéines « Z-disc » (ACTN2, MYOZ2) et des protéines de gestion du calcium. Une forme rare de CMH clinique d'apparition précoce est provoquée par des mutations du gène PRKAG2. Ce phénotype de CMH est aussi associé à une forme familiale de syndrome de pré-excitation (le syndrome WPW familial). Chez l'enfant, la CMH peut apparaître sous forme syndromique, dans le cadre, par exemple, d'un syndrome de Noonan, d'un syndrome de LEOPARD, de l'ataxie de Friedreich (AR), ainsi que de maladies métaboliques et neuromusculaires héréditaires6.
Cardiomyopathie dilatée (CMD)
La cardiomyopathie dilatée non ischémique se caractérise par une dilatation pathologique des cavités ventriculaires et par une diminution de la contractilité en l'absence de charge anormale. L'incidence est estimée à 5 à 8 personnes sur 100 000 et la prévalence chez l'adulte est de 1 sur 2 5007.
Bien que la cause reste souvent idiopathique, il est présumé que la moitié des cas de CMD correspondent à une forme familiale. Lorsqu'en plus d'une cardiomyopathie dilatée non ischémique, des troubles de la conduction AV surviennent également, le risque que la cardiopathie ait une origine génétique est plus élevé. À ce jour, on a identifié plus de 40 gènes impliqués dans la pathogenèse de la CMD familiale.
Récemment, le gène « titin » (TTN) a été identifié comme cause de la CMD familiale, ce qui permet désormais de rendre compte d'environ 25 % des cas de CMD idiopathique8. Le TTN est le plus grand gène présent dans le génome humain, ce qui explique qu'un grand nombre de variants de forme bénigne sont également retrouvés. L'interprétation correcte des variants TTN demeure un défi. À ce jour, seules des mutations tronquées (condons stop et « out-of-frame splice site ») ont été rapportées, et l'évaluation des mutations ponctuelles fait encore l'objet d'études scientifiques.
La présentation de la CMD est spécifique au gène concerné. Des mutations au niveau du gène « Phospholambane » (PLN) sont à l'origine d'une forme familiale de CMD, qui évolue à un jeune âge vers une défaillance de la fonction de pompe systolique9 ; par ailleurs, des mutations au niveau du gène « Lamin A/C » (LMNA/C) ont été associées à une forme particulièrement maligne de CMD, qui apparaît à un stade précoce, avec une bradycardie résultant de troubles de la conduction AV et une arythmie maligne. L'évolution vers une insuffisance cardiaque systolique apparaît généralement plus tardivement, à partir de la quarantaine. Les patients atteints d'une laminopathie ont généralement un mauvais pronostic en première phase, du fait de l'arythmie maligne, et par la suite en raison de l'insuffisance cardiaque terminale. Aujourd'hui, des facteurs de risque spécifiques aux gènes ont été définis pour les laminopathies, ce qui permet d'améliorer la prévention de la mort cardiaque subite dans cette population spécifique10.
La CMD liée au chromosome X est le résultat de mutations de la dystrophine combinées à une dystrophie musculaire (maladies de Becker et de Duchenne) ; en outre, chez les patients de sexe masculin, elle peut aussi apparaître de façon isolée, en l'absence de myopathie périphérique manifeste11.
La cardiomyopathie arythmogène (CMA)
La cardiomyopathie arythmogène se caractérise par l'infiltration épicardique progressive de tissu fibreux et lipomateux du myocarde. Ce processus conduit au déclin progressif de la fonction systolique et à des modifications de la conduction normale entraînant une instabilité électrique, généralement détectable en premier lieu au niveau du ventricule droit. La CMA est une maladie qui touche le système de l'adhérence, responsable de l'adhésion des cardiomyocytes entre eux ; en cas d'anomalies génétiques, elle touche le plus souvent les deux ventricules, mais des cas d'atteinte du ventricule droit ou du ventricule gauche principalement ont également été décrits. L'idée que la CMA toucherait uniquement le ventricule droit (une pathologie désignée sous le nom de ARVC) est désormais abandonnée.
La prévalence de la CMA est estimée à 1 personne sur 2 000 - 5 000. La CMA est fréquemment causée par des anomalies génétiques désmosomales (PKP2, DSP, DSG2, DSC2, JUP) et, moins fréquemment, par des mutations extra-desmosomales (notamment RYR2, DES, TMEM43, TGFB3, FLNC et TTN). La CMA se transmet principalement selon un mode autosomique dominant, mais de rares formes autosomiques récessives, telles que la maladie de Naxos reposant sur des mutations du gène JUP, sont également connues12. Un nouveau groupe récent de CMA, caractérisées par une captation importante, correspond au groupe des mutations FLNC (Filamine C), qui peuvent provoquer une forme arythmogène à la fois de la cardiomyopathie hypertrophique et de la cardiomyopathie dilatée.13
La CMA évolue en trois phases : une première phase asymptomatique, suivie d'une seconde phase arythmogène qui se manifeste le plus souvent chez l'adulte jeune, puis d'une troisième phase caractérisée par l'apparition d'une insuffisance cardiaque systolique. L'expression clinique au sein des familles est extrêmement variable et imprévisible. La CMA est une cause majeure d'événements malins, et est considérée comme responsable de 30 % des cas de mort cardiaque subite chez les athlètes. Il est généralement admis que la myocardite peut jouer un rôle dans la pathogenèse et qu'une lourde charge physique peut favoriser la progression de la CMA. Récemment, des arguments ont été mis en évidence en faveur de mécanismes oligogéniques, ce qui pourrait expliquer la corrélation hétérogène génotype-phénotype dans la CMA14. Le diagnostic clinique de la CMA est compliqué et repose sur un ensemble d'indications : observations structurelles, tracé d'ECG, observations moléculaires et histologiques qui, réunies sous une définition des critères du groupe d'étude, sont associées à une probabilité de diagnostic de CMA15.
La cardiomyopathie restrictive (CMR)
La CMR et une maladie rare caractérisée par un durcissement progressif de la paroi cardiaque empêchant la relaxation diastolique, et ce en présence de volumes systoliques et/ou diastoliques normaux (à faibles) et en l'absence d'une hypertrophie significative. À un stade plus tardif, la CMR évolue généralement vers une insuffisance cardiaque systolique.
La CMR primaire est causée par une troponinopathie et une desminopathie. La troponinopathie se caractérise par un risque arythmogène accru, tandis que la desminopathie se manifeste essentiellement par des troubles de la conduction AV et par une myopathie périphérique.16
La CMR secondaire familiale est le résultat d'une infiltration myocardique, comme dans l'amylose à TTR, ou peut résulter d'une accumulation anormale, comme dans l'hémochromatose et la maladie de Fabry.
Les cardiopathies électriques primitives
Contrairement aux cardiomyopathies, dans le cas des cardiopathies électriques primitives, les arythmies cardiaques menaçant le pronostic vital apparaissent en l'absence d'anomalies cardiaques structurelles détectables. Ce groupe inclut les pathologies suivantes : le syndrome du QT long (LQTS), le syndrome du QT court (SQTS), le syndrome de Brugada (SB), le syndrome de repolarisation précoce (ERS), la tachycardie ventriculaire polymorphe catécholaminergique (CPVT), la fibrillation ventriculaire idiopathique (FVI) et les troubles progressifs de la conduction cardiaque (PCCD).
Ces pathologies sont causées par des anomalies moléculaires dans les canaux ioniques intervenant dans le potentiel d'action cardiaque. Dans la plupart des cas, l'instabilité électrique résulte d'un renforcement ou d'une perte de la fonction de l'un des flux d'ions cardiaques, ce qui perturbe l'équilibre normal entre les forces dépolarisantes et repolarisantes. Il en résulte un allongement ou un raccourcissement anormal du potentiel d'action, et parfois un ralentissement anormal de la conduction, entraînant une augmentation de la dispersion du potentiel d'action local ou régional, une situation qui peut être qualifiée de pro-arythmogène. La majorité de ces affections sont provoquées par des mutations des gènes qui codent pour la sous-unité α des canaux ioniques cardiaques, tels que KCNQ1, KCNH2, SCN5A, CACNA1C, KCNJ2, KCNJ8, KCNJ11, KCND3, TRPM4, SLC4A3 et RYR2. D'autres mutations, moins fréquentes, affectent les sous-unités β et les protéines auxiliaires de ces flux d'ions, p. ex. CAV3, AKAP-9, SNTA-1, GPD1L, ABCC9, KCNE3, CASQ2, CALM1, CALM2, TRDN, DPP6, MOG1, SLMAP, CACNA2D1, KCNE1, KCNE2, SCNB1 et SCNB3.
Ici aussi, on observe donc une hétérogénéité génétique importante. Plusieurs gènes sont à l'origine d'une même pathologie, lorsqu'un effet similaire en termes mécanistiques est induit sur le potentiel d'action. Nous savons par exemple aujourd'hui que 16 gènes distincts peuvent provoquer individuellement une forme de LQTS. A contrario, selon la topologie et le type de mutation, des mutations au niveau d'un même gène peuvent induire des pathologies qui diffèrent énormément en termes phénotypiques lorsque l'effet mécanistique sur le coeur diffère. Des mutations du gène SCN5A peuvent induire un LQTS3, tandis que d'autres mutations de ce gène peuvent donner lieu à un SB, certaines mutations entraînant une cardiomyopathie dilatée et d'autres encore un trouble progressif de la conduction de type maladie de Lenegre-Lev (PCCD Type IA).
La fonction des canaux ioniques cardiaques est influencée par des facteurs tels que la température, l'équilibre électrolytique, l'équilibre hormonal et le PH. Par ailleurs, il existe une interaction pharmacogénétique complexe avec des médicaments susceptibles de renforcer ou d'affaiblir l'expression d'anomalies génétiques, les variations génétiques se produisant dans le canal ionique luimême influençant aussi l'affinité et la cinétique de l'interaction pharmacologique. Une interaction complexe se développe ainsi entre des facteurs acquis et héréditaires, qui exercent tous une influence sur la stabilité électrique du coeur déterminée selon un modèle polygénique. Ces interactions sont donc tout sauf prévisibles. Pour cette raison, il est indispensable d'exclure toujours une cause secondaire lorsqu'une cardiopathie électrique primitive, telle qu'un LQTS, SQTS ou SB, est suspectée.
Le diagnostic des cardiopathies électriques primitives est souvent difficile, car les anomalies diagnostiques sont souvent subtiles ou présentes de façon transitoire. De plus, au sein des familles présentant des cardiopathies électriques primitives, l'expression clinique est très variable et la pénétrance est souvent limitée.
Le syndrome du QT long (LQTS)
Le syndrome du QT long est causé par l'allongement pathologique du potentiel d'action cardiaque, ce qui augmente la dispersion transmurale. Le diagnostic est établi après l'exclusion de causes secondaires sur la base d'un allongement excessif récurrent du QTc, un état qui prédispose aux torsades de pointes, et à la mort cardiaque subite. La prévalence du syndrome du QT long est estimée à environ 1 personne sur 2 500 à 5 000. À ce jour, on distingue 16 génotypes distincts de LQTS ; 75 % des formes héréditaires du LQTS sont dues à des mutations des gènes KCNQ1 (LQST1), KCNH2 (LQST2) et SCN5A (LQTS3). Les formes de LQTS autosomiques dominantes étaient auparavant désignées sous le nom de « syndrome de Romano-Ward ». Une forme récessive rare, caractérisée par une surdité congénitale en plus d'un LQTS, repose sur des mutations de KCNQ1 ; ce trouble est connu sous le nom de « syndrome de Jervell Lange-Nielsen » (JLN)17.
La forme de LQTS acquise et réversible, fréquemment observée, résulte le plus souvent de troubles de l'équilibre électrolytique ou d'une interaction pharmacologique avec des médicaments allongeant l'intervalle QT. Pour consulter une liste de médicaments allongeant l'intervalle QT, veuillez consulter le site www.crediblemeds. org. Dans 10 à 15 % des cas des formes soi-disant « acquises » de LQTS, une prédisposition génétique sousjacente peut malgré tout être mise en évidence.
Le syndrome du QT court (SQTS)
Le syndrome du QT court est une maladie rare caractérisée par un raccourcissement excessif du potentiel d'action, ce qui se traduit à l'ECG par des valeurs QTc inférieures à 330 ms, souvent en association avec une non-adaptation de l'intervalle QT durant la bradycardie. Le SQTS cause, avec une haute pénétrance, un risque fortement accru de TV/FV polymorphe, la mortalité étant de 50 % avant l'âge de 40 ans. À ce jour, on estime que 8 gènes (KCNQ1, KCNH2, KCNJ2, CACNA1C, CACNB2, CACN2D1, SLC22A5 et SLC4A3) sont responsables de ce syndrome, qui se transmet sur un mode autosomique dominant18.
Le syndrome de Brugada (SB)
Le syndrome de Brugada repose sur des anomalies génétiques qui influencent à ce point la phase précoce du potentiel d'action que des anomalies de conduction épicardiques locales, généralement transitoires, apparaissent ; ces dernières sont de nature pro-arythmogènes et prédisposent à la FV et à la mort cardiaque subite. La mort cardiaque subite survient le plus souvent au repos entre la 3e et la 4e décennie et touche plus fréquemment les hommes que les femmes. Le diagnostic est établi sur la base d'anomalies à l'ECG de type 1 dans les dérivations précordiales droites, et rarement dans les dérivations inférieures.
La prévalence est estimée à 1 personne sur 2 000 ; toutefois, dans certaines régions telles que l'Asie du sud-est, le SB est nettement plus fréquent. Le SB se transmet sur le mode autosomique dominant.
Dans le passé, 23 gènes associés à la pathogenèse du SB ont été décrits. Des publications récentes montrent qu'il est possible que seul le gène SCN5A ait un impact clinique suffisamment important pour pouvoir provoquer à lui seul un syndrome de Brugada19. On suppose aujourd'hui que la pathogenèse du SB présente une architecture plus oligogène, car la corrélation génotype-phénotype au sein des familles n'est généralement pas linéaire et que les tests génétiques ne peuvent rendre compte que de 18 à 32 % des cas de SB sur le plan moléculaire. Les autres gènes jouent peut-être un rôle dans certaines familles, mais plutôt alors dans le contexte d'une pathogenèse multifactorielle plus complexe. De nouveaux scores de risque polygéniques ont été élaborés afin de résoudre le problème lié à la corrélation génotype-phénotype et à la stratification de risque problématique qui en résulte.
On rencontre également une forme de SB acquise induite par la médication. À nouveau, il existe un large éventail de médicaments susceptibles d'interagir avec les canaux ioniques et de favoriser les arythmies en cas de SB préexistant, ou d'induire une nouvelle forme réversible de SB (phénocopie). Quant à savoir si une prédisposition génétique sousjacente est également présente dans la phénocopie acquise du SB, la question n'a pas été tranchée. La liste de ces médicaments peut être consultée sur le site web suivant : www.brugadadrugs.org.
Le syndrome de repolarisation précoce (ERS)
L'ERS associé à des ondes J marquées à l'ECG est généralement un phénomène bénin couramment observé : jusqu'à 1 à 2 % de la population générale et jusqu'à 35 % chez les athlètes en bonne santé. Dans de rares cas, une élévation du point J (inférieure et/ou latérale) ≥ 2 mm a été liée à une mort cardiaque subite. À ce jour, 7 gènes (KCNJ8, CACNA1C, CACNB2, CACN2D1, ABCC9, SCN5A et SCN10A) ont été associés à la forme maligne de l'ERS.
La tachycardie ventriculaire polymorphe catécholaminergique (CPVT)
La CPVT est une maladie rare caractérisée par l'apparition d'une tachycardie ventriculaire polymorphe ou une tachycardie bidirectionnelle dans des situations de tonus adrénergique accru, p. ex. en cas d'effort ou d'émotion intense. Les arythmies apparaissent sous l'effet d'une surcharge intracellulaire en calcium dans le cardiomyocyte, donnant lieu à des post-dépolarisations tardives qui, à leur tour, peuvent induire une arythmie maligne.
La prévalence du syndrome est estimée à 1 personne sur 10 000. L'ECG au repos ne présente aucune anomalie caractéristique. Toutefois, lorsque la fréquence cardiaque augmente au-delà de 100-120 spm, un nombre croissant de contractions ventriculaires prématurées (PVC) multifocales apparaissent, qui deviennent progressivement plus polymorphes et prolongées lorsque la fréquence cardiaque continue à s'accélérer. Dans de nombreux cas, on assiste à l'apparition d'une tachycardie ventriculaire bidirectionnelle, qui se caractérise par une rotation de 180° autour de l'axe et se produit après chaque complexe ventriculaire. Cette arythmie peut dégénérer en FV et en mort cardiaque subite.
À ce jour, 6 gènes responsables de la CPVT ont été identifiés (RYR2, CASQ2, TRDN, CALM1, CALM2, CALM3), qui permettent d'expliquer environ 60 % des cas de CPVT en termes moléculaires20. La CPVT se transmet sur un mode autosomique dominant, à l'exception de la CPVT reposant sur des anomalies au niveau des gènes CASQ2 et TRDN, qui présente une transmission autosomique récessive.
La pénétrance du phénotype CPVT (syncope, arythmie maligne et/ou mort cardiaque subite) est estimée à 80 % avant l'âge de 40 ans. Il est donc toujours important d'identifier et de traiter tous les patients asymptomatiques afin de prévenir la survenue de ces événements.
La fibrillation ventriculaire idiopathique (FVI)
La FVI est un nom générique désignant des pathologies donnant lieu à une mort cardiaque subite sous l'effet d'une fibrillation ventriculaire, sans cause réversible démontrable ou cardiopathie structurelle ou électrique sous-jacente. L'ECG de base ne présente généralement aucune anomalie et la FV est généralement induite par des PVC monofocales à couplage court. Du fait de l'origine monofocale de ces PVC liées au réseau de Purkinje, l'ablation permet de réduire la charge d'arythmie de façon significative. Il est donc essentiel de surveiller à long terme les patients à risque de mort cardiaque subite afin de détecter ce type de PVC à couplage court qui induisent souvent de courtes salves avec des arythmies plus complexes. Dans les formes familiales de FVI, des mutations ont été retrouvées dans les gènes SCN5A et DPP621.
Tests en cascade
Le but de la cardiogénétique et de la prise en charge de la mort cardiaque subite est le traitement optimal de la maladie sousjacente et la prévention de récidives chez le proband. Par ailleurs, nous souhaitons identifier les membres de la famille à haut risque afin de mettre en place des mesures de prévention et/ou un traitement approprié si besoin.
L'examen cardiogénétique débute toujours chez le proband, supposé présenter l'expression la plus marquée de la maladie causale sous-jacente. Il est donc présumé que la corrélation génotype-phénotype est la plus manifeste chez le proband. Avant de détailler le génotype, il est dès lors crucial de caractériser de la façon la plus complète possible le phénotype observé chez le proband. Il servira à orienter la stratification du risque future et à aider à interpréter les données génétiques. Sur la base du profil génotype-phénotype obtenu, les parents de premier degré pourront ensuite faire l'objet d'examens, susceptibles de donner lieu à des examens chez les parents de deuxième degré, et ainsi de suite.
Chaque examen cardiogénétique doit donc avant tout être motivé par la présence d'un phénotype. Outre la stratification du risque, l'examen cardiogénétique chez les membres de la famille peut aussi aider à mieux cerner le phénotype familial et l'hétérogénéité clinique.
Compte tenu de l'hétérogénéité clinique, de la pénétrance incomplète et des incertitudes qui continuent parfois à planer en matière de symptômes et de pronostic dans les maladies cardiogénétiques, la décision de procéder à d'autres examens familiaux doit toujours être évaluée de manière critique au regard des dommages potentiels pour les personnes en termes d'impact sur la vie, l'anxiété et la résilience psychologique22. Cet aspect devient extrêmement important lorsque les examens concernent également de jeunes enfants. Cette évaluation complexe doit dès lors toujours être réalisée dans le cadre d'équipes de soins pluridisciplinaires, consistant la plupart du temps en un(e) cardiologue clinique spécialisé( e) en affections cardiaques héréditaires, un(e) généticien(ne) clinique, un(e) généticien(ne) moléculaire, un(e) conseiller(-ère) génétique, un(e) psychologue et un(e) infirmier(-ère) spécialisé(e) en cardiogénétique.
Phénotypage
L'examen cardiovasculaire doit comprendre au minimum une anamnèse personnelle et familiale détaillée, un examen clinique, un ECG au repos, un ECG à l'effort, une échocardiographie et un enregistrement Holter sur 24 heures avec surveillance QT/QTc. Si nécessaire, ces examens peuvent être complétés par des examens spécifiques à la maladie, comme une RMC, une TDM cardiaque, un test à l'ajmaline, un SAECG, une échocardiographie à l'effort et une investigation électrophysiologique. Ensuite, un arbre généalogique est élaboré afin de mieux définir le phénotype familial et d'identifier d'autres membres de la famille susceptibles de fournir des informations.
Lors de l'anamnèse, une attention particulière doit être portée aux symptômes d'alarme et le patient sera interrogé en profondeur sur les événements malins subis, car ces informations peuvent fournir des indications importantes sur la cause.
Symptômes d'alarme
Le symptôme qui oriente par excellence vers une arythmie maligne est la syncope cardiogénique. Il s'agit d'un événement syncopal qui apparaît de façon très brutale sans prodromes et qui entraîne une perte de connaissance complète de courte durée. Après quelques secondes, le patient reprend généralement complètement conscience. Il n'y a pas de phase post-ictale. La survenue d'une incontinence est un signe d'atonie musculaire résultant de l'hypotension ; elle ne contricontribue pas au diagnostic différentiel de l'événement. Une syncope brutale constitue un symptôme d'alarme qui nécessite toujours un examen cardiaque complémentaire.
Les syncopes qui surviennent pendant (et non après) l'effort ou en position allongée sont également toujours suspectes pour un événement cardiaque. Il est important d'interroger le patient de façon proactive à ce sujet durant l'anamnèse, car les patients n'associent pas toujours une perte de connaissance subite à des problèmes cardiaques. Souvent, le patient dit qu'il est « tombé dans les pommes » ou a eu un « trou noir », et l'événement est ensuite ignoré.
Des palpitations symptomatiques associées à une pré-syncope indiquent la présence d'une arythmie dotée d'un impact hémodynamique significatif. Cela peut être la conséquence tant d'arythmies supraventriculaires que ventriculaires, et cela nécessite toujours un examen complémentaire ciblé.
La fibrillation auriculaire, l'angor et l'insuffisance cardiaque au début précoce peuvent être des marqueurs, respectivement, d'une instabilité électrique accrue telle qu'observée dans le SB et le LQTS, d'une ischémie relative dans la cardiomyopathie hypertrophique et d'une insuffisance cardiaque résultant d'une cardiomyopathie sous-jacente.
Chez les patients qui présentent une épilepsie inexpliquée jamais documentée par EEG, une syncope épileptiforme se révèle parfois être la conséquence d'une arythmie maligne. Chez l'enfant, les absences peuvent être le résultat d'une bradycardie anormale, telle que parfois observée dans le SB.
L'hyperventilation est le plus souvent associée à de l'anxiété, de la détresse subjective et des palpitations cardiaques. Dans certains cas, la crise d'hyperventilation est déclenchée par des palpitations symptomatiques.
La mort subite
En cas de mort cardiaque subite, il est important de vérifier dans quelles circonstances l'événement est survenu. Si possible, les témoins seront contactés, car ils peuvent souvent fournir de précieuses informations sur le déroulement de l'événement et sur les circonstances.
Ensuite, il est très important de demander à obtenir le premier rythme documenté au service des urgences ayant pratiqué la réanimation. Par ailleurs, lorsqu'un DEA a été utilisé, une interprétation de l'événement peut être demandée. La documentation de torsades de pointes ou d'une TV bidirectionnelle est assez pathognomonique d'un LQTS et d'un CPVT, respectivement.
En cas d'arythmies récurrentes, la fréquence de base et le tracé initial ainsi que l'intervalle de couplage (p. ex. PVC à couplage court vs tracé initial dépendant de la pause, séquence court-long-court, dégénérescence progressive des PVC en pTV/FV) peuvent fournir des indications complémentaires sur la cause. Dès lors, il est particulièrement recommandé d'utiliser des dispositifs de surveillance éventuels tels qu'un moniteur cardiaque implantable (ILR), un stimulateur cardiaque ou un ICD.
L'autopsie
Lorsque le patient est décédé, il est important de pratiquer une autopsie. L'autopsie peut en effet révéler des informations importantes sur la cardiopathie structurelle sous-jacente (ou sur son absence complète) et exclure des causes toxicologiques ; elle permet en outre d'obtenir un échantillon d'ADN pour analyses génétiques. Lorsque le patient n'est pas décédé en milieu hospitalier, un échantillon d'ADN ne peut être prélevé qu'avant l'établissement de l'acte de décès. Toutefois, les équipes de soins d'urgence peuvent être formées à conserver le tube endotrachéal ou l'accès intra-osseux en tant qu'échantillon post-mortem à envoyer au centre cardiogénétique pour extraction et analyse de l'ADN.
Ici aussi, les dispositifs de surveillance du rythme sont particulièrement indiqués pour déterminer la cause arythmogène de la mort cardiaque subite. Dans la pratique, tout cela se produit trop rarement, de sorte que la seule possibilité d'information structurelle et de recherche génétique est souvent perdue. Cela crée une situation dans laquelle toute information phénotypique est absente.
Le phénotype SUDS
Lorsque le patient est décédé des suites de l'événement et qu'aucune cause démontrable ne peut être détectée, on parle de mort subite de cause inconnue ou SUDS (sudden unexplained death syndrome). Souvent, aucune autopsie n'a été pratiquée ou l'autopsie n'a pas pu expliquer la cause du décès.
Ce phénotype particulier entrave les options de prévention familiale à moins qu'un échantillon de tissu ou de sang n'ait été prélevé lors de l'autopsie. Dans ce cas, une autopsie moléculaire peut encore être réalisée, laquelle peut révéler le diagnostic et nous permettre d'identifier et de protéger d'autres membres de la famille à haut risque.
Génotypage
Après un phénotypage approfondi chez le proband, un échantillon de sang EDTA est prélevé pour analyse génétique. Lors de l'autopsie, un échantillon de tissu, généralement du foie, est envoyé pour être soumis à une recherche génétique.
Diagnostic
Le but de l'examen génétique est de confirmer le diagnostic chez le proband lorsqu'un phénotype clair est présent. Lorsque le phénotype n'est pas clair, ou quand on ne reconnaît aucun phénotype, l'examen cardiogénétique diagnostique peut aider à établir le diagnostic.
Même en cas de décès de type SUDS (sans cause identifiable) et lorsque l'autopsie n'a pas été réalisée ou était négative, le génotypage post-hoc ou l'autopsie moléculaire peut encore aider à poser le diagnostic. Cette technique spécialisée permet de poser un diagnostic chez environ 25 % des morts subites non résolues chez des jeunes, de manière à mieux protéger les membres de la famille à haut risque.
Une fois qu'une mutation pathogène est identifiée, il est facile de déterminer avec certitude et indépendamment de la présence de symptômes si un membre de la famille est porteur du même facteur pathogène. Cette recherche génétique prédictive permet ainsi de prédire à un stade pré-symptomatique s'il est possible que le phénotype s'exprime chez la personne. De cette manière, l'examen cardiogénétique permet de suivre des patients à un stade présymptomatique, de prendre les mesures préventives primaires nécessaires pour retarder/ prévenir la manifestation de la maladie, et le cas échéant, de détecter le plus tôt possible les signes pathologiques et/ou les symptômes (d'alarme) précoces.
De plus, il est possible d'empêcher qu'une mutation pathogène ayant un impact phénotypique potentiellement important soit transférée à une génération suivante future. Via la fécondation in vitro, le test génétique préimplantatoire (PGT - Pre-implantation Genetic Testing), qui consiste à tester une cellule d'embryon au stade précoce pour vérifier si elle est porteuse, offre la possibilité de replacer uniquement l'embryon qui n'est pas porteur de la mutation pathogène. Cela empêche la transmission et la maladie, et celle-ci peut ainsi être stoppée dans la descendance familiale. Dans certaines publications, cette procédure est parfois appelée prévention primordiale.
Enfin, le génotypage nous permet de mieux différencier les syndromes les uns des autres. Par exemple, le groupe de patients encore rassemblés actuellement sous le diagnostic de travail « cardiomyopathie dilatée idiopathique » diminuera progressivement. Nous savons à présent qu'il existe aussi entre autres les cardiomyopathies arythmogènes, les laminopathies, les desminopathies ainsi que la cardiomyopathie due à une mutation du phospholambane ou du titin : ces pathologies cardiaques existent chacune avec leur évolution naturelle et avec des besoins spécifiques de prévention et de traitement. Pour cette dernière raison, nous évoluerons vers un diagnostic moléculaire spécifique au gène pour certaines de ces affections, comme pour le LMNA/C, DES et PLN où c'est déjà le cas.
Prévention
En plus de générer des options de prévention primaire et primordiale, la recherche cardiogénétique facilite dans certains cas la décision quant aux mesures de prévention secondaire immédiate. Il n'est pas rare que des symptômes d'alarme tels que les syncopes cardiogéniques ne soient pas reconnus comme tels et ils sont donc souvent ignorés par les patients. Si un tel symptôme d'alarme, associé au diagnostic cardiogénétique, s'avère représenter un risque fortement accru de mort cardiaque subite, un défibrillateur automatique implantable (ICD - Implantable Cardioverter Defibrillator) peut être nécessaire pour protéger le patient d'une mort cardiaque subite.
Les progrès rapides de nos techniques cardiogénétiques et l'élaboration de registres et de bases de données à l'échelle mondiale montrent également de plus en plus clairement que le génotypage peut aussi contribuer directement à la stratification du risque chez le proband et ses proches. Au niveau du gène et en association avec d'autres facteurs de risque, nous savons que les mutations de décalage de cadre (frameshift) et les mutations de troncature dans le gène LMNA/C entraînent un risque de mort subite supérieur aux mutations ponctuelles, raison suffisante pour que les porteurs masculins envisagent l'implantation d'un ICD10.
Dans de rares cas, le génotype associé à l'expression familiale est déjà suffisant, comme dans la tachycardie ventriculaire polymorphe catécholaminergique (CPVT) atypique où les patients atteints ne présentent aucun phénotype démontrable quel que soit le test clinique, le statut de porteur de la mutation familiale RYR2 va de pair avec une incidence extrêmement élevée d'événements de mort subite dans la famille, ce qui signifie qu'être porteur de la mutation pathogène peut justifier en soi l'implantation d'un ICD.
Entretemps, il a également été démontré que certaines mutations spécifiques comportent un risque extrêmement élevé, p. ex. la mutation p.Arg92Gln (ou p.Arg102Gln) dans le gène TNNT2 qui provoque une forme maligne de cardiomyopathie hypertrophique (CMH) caractérisée par une mortalité de 50 % avant l'âge de 50 ans23. Il va sans dire que, bien que notre stratification classique du risque pour la CMH n'en tienne pas compte et que, dès lors, nos critères de remboursement n'aient pas été adaptés, le statut de porteur de ce génotype devrait en lui-même être suffisant pour justifier l'implantation d'un ICD afin de prévenir une mort cardiaque subite. Ainsi, la collaboration entre les centres de cardiogénétique a permis d'identifier récemment une mutation fondatrice belge dans le gène SCN5A causant une forme plus maligne du syndrome de Brugada24.
Traitement
Le génotypage peut également avoir un impact direct sur la prise en charge des patients. Au niveau du gène, nous savons qu'un syndrome du QT long de type 1 ou LQTS1 basé sur des mutations KCNQ1 sera traité autrement qu'un LQTS2 reposant sur des mutations KCNH2 et qu'un LQTS3 résultant de mutations SCN5A, puisque les canaux ioniques de chacun de ces gènes montrent une expression fonctionnelle spécifique différente dans la cascade du potentiel d'action cardiaque. Par exemple, dans le LQTS1, des troubles du rythme se produisent davantage à des fréquences cardiaques rapides, alors que c'est moins le cas dans le LQTS2, et que dans le LQTS3, ces troubles s'observent plutôt à des fréquences lentes.
Entretemps, la cardiogénétique a également évolué vers une thérapie spécifique à la mutation. Des mutations dites de « gain de fonction » (gain-of-function) dans le gène SCN5A, comme la mutation p.Arg222Gln qui renforce excessivement la fonction du canal sodique, peuvent susciter une hyperexcitabilité entraînant très fréquemment une extrasystolie. Cela donne souvent lieu à une forme de tachycardiomyopathie dès le jeune âge, qui aboutit à une insuffisance cardiaque. Cette nouvelle entité est appelée « contractions prématurées multifocales ectopiques dépendantes de Purkinje » ou en abrégé MEPPC (multifocal ectopic Purkinje- related premature contractions)25. De notre propre expérience, nous savons que la thérapie par ablation de ces extrasystoles est impossible en raison de l'aspect multifocal extrême en tant que signe du myocarde hyperexcité. En raison du risque de pro-arythmie, il est contre-indiqué d'utiliser des antagonistes du calcium de classe Ic selon la classification de Vaughan Williams tels que la flécaïnide chez les patients qui présentent une diminution de la FEVG. Toutefois, il existe ici une translation évidente d'un génotype avéré de type « gain de fonction » lié au canal sodique à un phénotype d'hyperexcitabilité avec tachycardiomyopathie secondaire. Le traitement par un antagoniste du calcium agit directement sur ce mécanisme pathogène, réduit la fonction génétique renforcée du canal sodique, élimine l'hyperexcitabilité et conduit chez ces patients au rétablissement complet de la fonction du VG dans un délai de trois mois, ainsi qu'à la disparition de tous les symptômes. À nouveau, il s'avère que le génotypage permet d'affiner le traitement pour des patients particuliers et que les anciens paradigmes sont sans doute trop généraux pour la cardiologie moléculaire translationnelle amenée à voir le jour dans un avenir proche.
Qu'est-ce qu'une mutation pathogène ?
Le génome humain comprend quelque 21 000 gènes. Des variations peuvent intervenir dans chacun de ces gènes. Des années durant, on a pensé qu'une variation génétique dans un gène était automatiquement la cause de la maladie pour laquelle on tentait de mettre en évidence une cause génétique.
Aujourd'hui, on sait que les anomalies génétiques peuvent relever de la variation évolutive normale et qu'elles ne sont pas toujours pathogènes. De manière générale, les variants génétiques observés fréquemment au sein de la population, appelés « polymorphismes », sont considérés comme bénins. La survenue des variants génétiques dans une population témoin est appelée « réquence de l'allèle mineur » (minor allel frequentie, ou MAF). La survenue fréquente d'un variant dans la population générale (MAF élevée > 1 %) est considérée comme un signe que ce variant a été mieux toléré au cours de l'évolution et n'a donc sans doute eu aucun impact pathogène majeur, raison pour laquelle il a pu se développer et se propager à une fréquence élevée. Inversement, le risque de pathogénicité d'un variant génétique rare est estimé plus élevé, car celui-ci a moins de chances de se transmettre sur le plan évolutif, a fortiori lorsque le phénotype donne lieu à un risque accru de mort cardiaque subite.
Il existe cependant des polymorphismes très fréquents qui ne provoquent pas la maladie en eux-mêmes, mais qui, associés à d'autres variants génétiques, peuvent amplifier ou affaiblir le phénotype. Ce type de variants, qui ne deviennent cliniquement significatifs qu'au sein d'une constellation spécifique de variations génétiques, portent le nom de variants de type « disease modifiers » ou « modificateurs de la maladie ».
Ces variants sont certainement importants pour les cardiopathies électriques, car le potentiel d'action cardiaque est formé par une cascade complexe de flux ioniques qui s'influencent mutuellement. Une modification mineure de l'expression fonctionnelle d'un composant peut avoir des répercussions sur un ou plusieurs autres composants. Il a ainsi été démontré sur le plan fonctionnel que deux variants, p.Lys897Thr et p.Ala1116Val, du gène KCNH2 codant pour le canal potassique IKr et qui ne possèdent en soi aucun potentiel pathogène, peuvent induire un LQTS lorsqu'ils surviennent en association. Le variant p.Lys897Thr du gène KCNH2 est présent chez 27 % de la population caucasienne. Les soi-disant polymorphismes peuvent donc bel et bien présenter des propriétés modificatrices de la maladie26. Et bien entendu, tout variant rare n'est pas automatiquement pathogène et n'intervient pas nécessairement dans le processus pathologique.
Par ailleurs, il apparait aussi d'emblée que les maladies génétiques ne présentent pas toujours une architecture monogénique, dans la mesure où il est fréquent que plusieurs variants génétiques de différents gènes interviennent dans le développement de l'expression du génotype-phénotype finale.
Il s'ensuit qu'un variant peut présenter des propriétés pathogènes chez un patient, alors que chez un autre individu ou membre de la famille, en fonction de la présence de variants génétiques apparemment banals, ce même variant n'induira absolument aucun phénotype. L'ensemble des facteurs génétiques susceptibles de jouer un rôle dans le développement d'une corrélation génotype-phénotype est désigné sous le nom de « contexte génétique », qui diffère fortement d'un individu à l'autre, et ce même au sein d'une même famille. D'autre part, nous connaissons des mutations pathogènes qui donnent lieu à un phénotype de maladie plus léger qui s'exprime en outre à un âge avancé, bien après l'âge reproductif, ce qui fait que le mécanisme de sélection évolutif n'a pas fonctionné et que ces mutations continuent à ségréger tranquillement au fil des générations. Ces mutations présentent donc une fréquence anormalement élevée au sein de la population27.
Apparaît donc ainsi un spectre de pathogénicité qui varie de variants banals à des mutations pathogènes en passant par des variants de type polymorphismes et des modificateurs de la maladie (disease modifiers). Le contexte génétique de chaque personne étant unique, l'évaluation de la pathogénicité des variants génétiques doit toujours s'effectuer sur une base individuelle et en faisant preuve d'une extrême prudence.
Classification des variants génétiques
À ce jour, les variants génétiques sont répartis en 5 classes en fonction de leur pathogénicité : bénigne (UVKL1), probablement bénigne (UVKL2), signification clinique indéterminée (UVKL3), probablement pathogène (UVKL4) et pathogène (UVKL5).
La classification des variants repose sur les recommandations de l'ACMG28 et tient compte de l'apparition du variant dans les populations témoins ainsi que dans les banques de données de séquences génétiques et spécifiques à la maladie, telles que LOVD, GnomAD et ExAC. Par ailleurs, le type de variation génétique est important, comme les mutations de type troncature, qui induisent des modifications importantes dans la protéine résultante et possèdent dès lors un plus grand potentiel pathogène que les mutations ponctuelles. En outre, la pathogénicité des variants génétiques est estimée à l'aide de la conservation philogénétique. Les positions des acides aminés fortement conservées à travers les différentes espèces s'avèrent être extrêmement importantes sur le plan évolutif. Les variants occupant ces positions sont donc estimés plus pathogènes. Enfin, sur la base d'algorithmes de prédiction « in silico », l'impact sur la structure primaire, secondaire et tertiaire de la protéine a été évalué en tant que mesure de pathogénicité.
Ce n'est que pour les variants probablement pathogènes (UVKL4) et pour les variants pathogènes (UVKL5) qu'une corrélation suffisante a été démontrée entre le génotype et le phénotype ; dès lors, seuls ces variants peuvent s'utiliser pour une recherche cardiogénétique prédictive chez les membres de la famille.
Les variants bénins (UVKL1) et probablement bénins (UVKL2) n'ont pas de signification clinique et ne sont dès lors pas rapportés.
Le groupe de variants de loin le plus large identifié à ce jour est composé des variants de signification clinique indéterminée (UVKL3), pour lesquels il existe des indications qu'ils pourraient intervenir dans l'étiologie du phénotype ; les données sont toutefois insuffisantes pour prouver ou réfuter un lien de causalité. Pour cette raison, les variants de classe 3 ne peuvent pas être utilisés pour des tests génétiques prédictifs de membres de la famille pré-symptomatiques. Dans certains cas, l'analyse de co-ségrégation peut contribuer à mieux expliquer la signification des variants de classe 3, ce qui permet de créer une nouvelle classification de classes de pathogénicité informatives 4 ou 5.
Indications de la recherche cardiogénétique
Équipe pluridisciplinaire
Les maladies cardiogénétiques nécessitant une prise en charge requérant de larges connaissances tant durant la vie que lors du décès, il est conseillé que la recherche cardiogénétique soit menée par des équipes pluridisciplinaires composées idéalement de cardiologues spécialisés dans les maladies cardiogénétiques chez l'adulte et l'enfant, des généticiens moléculaires et cliniques ainsi que des anatomo-pathologistes spécialisés en cardiopathies. Idéalement, l'équipe sera soutenue par des conseillers génétiques, des psychologues et un personnel infirmier. Une approche holistique spécialisée, mais large pourra ainsi être mieux garantie.
Conseils génétiques
L'obtention de conseils génétiques est fortement recommandée pour toutes les familles confrontées à une mort cardiaque subite lorsqu'une cause héréditaire est suspectée. De même, lorsque le patient est décédé et que la cause ne peut être identifiée, les conseils génétiques sont indispensables.
Ils permettent de recueillir les informations nécessaires sur les antécédents de la maladie, les symptômes d'alarme, le déroulement de l'événement du proband, mais aussi d'élaborer un arbre généalogique de la famille et d'avancer une hypothèse concernant la cause sousjacente éventuelle. Le phénotype et le phénotype familial éventuel sont ainsi décrits, afin de pouvoir réaliser une évaluation pondérée de l'intérêt, des avantages et inconvénients et des résultats attendus d'une analyse génétique.
À cet égard, il est toujours important d'offrir la possibilité d'un soutien psychologique au proband survivant et aux membres de sa famille.
Examen diagnostique
La suspicion d'une maladie cardiaque d'origine génétique en cas de mort cardiaque subite constitue une indication sérieuse pour réaliser un test génétique, afin d'identifier la cause moléculaire, mais aussi de permettre l'identification des membres de la famille à haut risque (pré-symptomatiques) par le test de portage reposant sur le dépistage en cascade. Chez les parents de premier degré d'un proband ayant subi une mort cardiaque subite avec suspicion de maladie cardiaque héréditaire, le phénotypage cardiaque est recommandé et, dans la mesure du possible, des tests de dépistage génétiques en cascade seront appliqués29.
En cas de mort cardiaque subite inexpliquée et lorsque la cause reste inexpliquée après une évaluation d'experts multidisciplinaire, mais que certaines indications suggèrent une cause arythmogène, il est recommandé de procéder à des tests génétiques ciblant des cardiopathies électriques primitives sous-jacentes. En cas de mort subite inexpliquée ou SUDS chez un patient jeune, un test génétique ciblant les cardiopathies électriques est envisageable.
Chez les parents de premier degré, il est recommandé de répéter le test de dépistage cardiaque tous les 3 à 5 ans, ou à intervalles plus rapprochés lorsque plus d'une mort cardiaque subite survient dans la famille.
Test prédictif
Le test de portage (pré)-symptomatique permet d'identifier les membres de la famille ayant une prédisposition au phénotype familial. Le test génétique prédictif est réservé aux variants probablement pathogènes et pathogènes avérés de classes 4 et 5.
Le test génétique préimplantatoire
La possibilité d'empêcher la transmission de mutations génétiques pathogènes est réservée aux variants probablement pathogènes (classe 4) et aux variants pathogènes (classe 5). Dans cette optique, un parcours de FIV est mis sur pied, avec un test de portage sur cellule unique, suivi d'une sélection embryonnaire.
Conclusion
La cardiogénétique est le pilier de la dimension moléculaire de la cardiologie, qui nous permet d'améliorer et d'individualiser encore plus en avant nos possibilités de stratification du risque, de prévention et de traitement. Bien que nous n'en soyons encore qu'à la première phase de cette nouvelle avancée dans nos connaissances et notre compréhension des cardiopathies, il s'agit désormais bel et bien de notre nouvelle réalité. Chacune des mutations citées plus haut a été identifiée dans la population belge. Malgré les progrès rapides enregistrés, de nombreux variants fournissent encore trop peu d'informations pour être utilisés en clinique. Il est probable que l'intelligence artificielle et l'analyse des « big data » nous aideront respectivement à obtenir un meilleur phénotypage et à améliorer l'analyse des variants, afin de pouvoir interpréter la signification spécifique des variants pour un patient donné dans un contexte polygénique.
Pour réaliser ces objectifs, les conditions suivantes doivent être remplies : prélever un échantillon d'ADN lors de tout décès suspect, développer une corrélation génotypephénotype correcte et mettre en place une approche pluridisciplinaire soutenue au sein de centres cardiogénétiques spécialisés, l'objectif principal étant d'améliorer la prévention de la mort cardiaque subite.
Références
- Hasselqvist-Ax, I. et al. Early cardiopulmonary resuscitation in out-ofhospital cardiac arrest. N Engl J Med, 2015, 372 (24), 2307-2315.
- Perkins, G.D. et al. European Resuscitation Council Guidelines 2021: Executive summary. Resuscitation, 2021, 161 (1).
- O'Mahoney, C. et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J, 2014, 35 (30), 2010-2020.
- Van der Werf, C. et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in The Netherlands. Heart Rhythm, 2010, 7 (10), 1383-1389.
- Hendrix, A. Sudden cardiac death in the young: incidence and consequences. University of Utrecht, 2011.
- Sabater-Molina, M. et al. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin Genet, 2018, 93 (1), 3-14.
- Jefferies, J.L. et al. Dilated cardiomyopathy. The Lancet, 2010, 375 (9716), 752-762.
- Herman, D.S. et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med, 2012, 366 (7), 619-628.
- Schmitt, J.P. et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science, 2003, 299 (5611), 1410-1413.
- van Rijsingen, I.A. et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol, 2012, 59 (5), 493- 500.
- Arbustini, E. et al. Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J Am Coll Cardiol, 2000, 35 (7), 1760-1768.
- Elliott, P.M. et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail, 2019, 21 (8), 955-964.
- Corrado, D. et al. Filamin C: A New Arrhythmogenic Cardiomyopathy-Causing Gene? JACC Clin Electrophysiol, 2018, 4 (4),515-517.
- Ye, J.Z. et al. Reevaluation of genetic variants previously associated with arrhythmogenic right ventricular cardiomyopathy integrating population-based cohorts and proteomics data. Clin Genet, 2019, 96 (6), 506-514.
- Markus FI et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J, 2010, 31 (7), 806-814.
- Arbustini, E. et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail, 2006, 8 (5), 477-483.
- Shimizu, W. et al. Genetics of long-QT syndrome. J Hum Genet, 2016, 61 (1), 51-5.
- Raschwitz, L.S. et al. Differences in Short QT Syndrome Subtypes: A Systematic Literature Review and Pooled Analysis. Front Genet, 2020, 10, 1312.
- Hosseini, S.M. et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation, 2018, 138 (12), 1195-1205.
- Wleklinski, M.J. et al. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J Physiol, 2020, 598 (14), 2817-2834.
- Postema, P.G. et al. Founder mutations in the Netherlands: familial idiopathic ventricular fibrillation and DPP6. Neth Heart J, 2011, 19 (6), 290-296.
- Manrai, A.K. et al. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med, 2016, 375 (7), 655-665.
- Ripoll-Vera, T. et al. Clinical and Prognostic Profiles of Cardiomyopathies Caused by Mutations in the Troponin T Gene. Rev Esp Cardiol (Engl Ed), 2016, 69 (2), 149-158.
- Sieliwonczyk, E. et al. Clinical characterization of the first Belgian SCN5A founder mutation cohort. Europace, 2020, euaa305.
- Laurent, G. et al. Multifocal ectopic Purkinjerelated premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol, 2012, 60 (2), 144-156.
- Crotti, L. et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation, 2005, 112 (9), 1251-1258.
- Buxbaum, J.N. et al. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med, 2017, 19 (7), 733-742.
- Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17 (5), 405-424.
- Stiles, M et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm, 2021, 18 (1), e1-e50.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.