Inleiding
Het plotse en vaak onverwachte overlijden van familieleden of kennissen is een gekend probleem dat niemand onberoerd laat. Wanneer het overlijden optreedt binnen het uur na het ontstaan van de symptomen, en het gevolg is van een hartziekte, spreken we van plotse hartdood. We kennen allemaal de berichten uit de media waarin het plotse overlijden meegedeeld wordt van vaak jonge, schijnbaar gezonde mensen dat benoemd wordt als 'hartfalen' of 'hart aderbreuk'. De precieze oorzaak blijft vaak onduidelijk en ook na autopsie kan vaak geen definitieve etiologie aangetoond worden, wat niet zelden uitmondt in de aspecifieke einddiagnose 'hartstilstand'.
Met een jaarlijkse incidentie van één tot twee overlijdens per duizend personenjaren heeft plotse hartdood een belangrijke maatschappelijke impact. In België komt dit neer op ongeveer 5-10 % van alle natuurlijke doodsoorzaken, wat goed is voor ca. 10 000 overlijdens per jaar.
Aangezien onze bevolking niet systematisch tot bekwame eerste hulpverleners opgeleid is, terwijl het uitblijven of laattijdig toekomen van omstander-CPR de overlevingskansen ernstig compromitteert, blijft de kans op overleving na een reanimatie slechts 4-10 % en bij succesvol herstel van spontane circulatie blijft de kans op restitutie zonder neurologische restschade erg beperkt1,2. Heel vaak gaat het om jonge actieve mensen waardoor de jaarlijkse socio-economische en maatschappelijke impact enorm is.
Ondanks onze uitgebouwde urgentienetwerken, de moderne medische kennis en laagdrempelige toegang tot hoogtechnologische cardiale zorg en ondersteuning, schijnen we plotse hartdood als klinische entiteit niet goed onder controle te krijgen. Onze cardiale diagnostiek is niet in staat om ziektebeelden afdoende te verklaren, waardoor we gewoon geworden zijn te werken met diagnoses zoals idiopathisch gedilateerde cardiomyopathie of idiopathische VF. Wellicht bevinden er zich achter deze idiopathische verzamelingen unieke aandoeningen die een meer specifieke aanpak vereisen en waar met gepaste maatregelen plotse hartdood beter kan vermeden worden. Ook onze op klassieke klinische indicatoren gebaseerde risicostratificatie lijkt een sterk verhoogd risico voor plotse hartdood niet steeds goed te kunnen inschatten. Daaruit volgt een preventiestrategie met ICD-implantatie waarna vaak geen aritmie meer gezien wordt, terwijl patiënten zonder ICD ondanks degelijke cardiale opvolging toch maligne events doormaken omdat ze als 'laag' risico ingeschat werden. Zo is het goed te beseffen dat we slechts de helft van de hoogrisico hypertrofe cardiomyopathiepatiënten beschermen wanneer we met de huidige risicostratificatie een drempel tot ICD-implantatie van 6 % 5-jaarsrisico voor plotse hartdood blijven hanteren3.
Bovendien worden in België bij een plotse hartdood zeer zelden autopsies uitgevoerd. Men gaat ervan uit dat het bij een natuurlijk overlijden geen zin meer heeft om de doodsoorzaak te achterhalen. Wanneer toch een autopsie gebeurt, blijft de doodsoorzaak door de afwezigheid van overte structurele hartafwijkingen vaak onbekend, wat uitmondt in 'plotse onverklaarde dood' (SUDS: Sudden Unexplained Death Syndrome). Wanneer bij een 'verdacht' overlijden een gerechtelijke autopsie gebeurt, is de obductie er uitsluitend op gericht om een criminele doodsoorzaak uit te sluiten, niet om een klinisch relevante doodsoorzaak op te sporen. Zowel bij civiele als gerechtelijke autopsies wordt niet routinematig een DNA-staal afgenomen en gaat de mogelijkheid tot post-hoc cardiogenetisch onderzoek bij de proband vaak onherroepelijk verloren. Ten slotte worden familieleden wanneer de proband overleden is slechts zelden verwezen voor verder cardiogenetisch onderzoek.
Kortom, er lijken waardevolle kansen verloren te gaan waardoor er een lange weg te gaan blijft om het management en de preventie van plotse hartdood te verbeteren. Nochtans werd het aangetoond dat tertiaire screening en management van plotse hartdood overlevenden en/ of hun familie kost-effectief kan zijn4. In dit artikel geven we een overzicht over de huidige kennis over plotse dood, de gekende oorzaken met de focus op de genetische aspecten, alsook de opportuniteiten en de hiaten in ons gezondheidssysteem om te komen tot betere plotse hartdood preventie.
Etiologie
Plotse hartdood is in 9 van de 10 gevallen het directe gevolg van maligne hartritmestoornissen ventrikeltachycardie (VT) en/of fibrillatie (VF) als 'final common pathway' van vooraf bestaand cardiaal lijden. Slechts zelden gaat het om acuut pompfalen of catastrofale structurele afwijkingen zoals ventrikelruptuur.
Vaak zijn er voorafgaand aan het plotsedoodevent helemaal geen symptomen of worden symptomen niet als cardiaal alarmteken herkend, waardoor patiënten vooraf meestal geen cardiaal onderzoek ondergaan. Hierdoor is in 30 % het plotse hartdood event het eerste en enige symptoom van de onderliggende hartziekte. Wanneer de patiënt de episode overleefd heeft, kan het klinisch cardiaal nazicht niet altijd hartafwijkingen detecteren. Ook wanneer de patiënt overleden is, wordt bij een autopsie vaak geen duidelijke oorzaak teruggevonden.
Algemeen is de onderliggende oorzaak van plotse hartdood in ca. 60-80 % van de gevallen toe te schrijven aan ischemisch hartlijden. Wanneer we dit verder onderverdelen naar leeftijd weten we dat in de leeftijdsgroep > 45 jaar de grote meerderheid van plotse hartdood het gevolg is van een acuut coronair syndroom (ACS).
Wanneer plotse hartdood voorkomt bij mensen < 45 jaar kan een onderliggende erfelijke oorzaak aangetoond worden in ca. 80 % van de gevallen5. Dit is belangrijk aangezien het plotse hartdood event bij de indexpatiënt hierdoor als een indicator beschouwd moet worden van een familiale hartziekte waarbij er mogelijks nog andere hoogrisicopatiënten in dezelfde familie aanwezig zijn die in aanmerking komen voor preventieve maatregelen. Hier ontstaat een belangrijke opportuniteit om tot betere preventie van plotse hartdood te kunnen komen. Het is dan ook van cruciaal belang de oorzaak van plotse hartdood bij de indexpatiënt of proband te achterhalen zodat de juiste preventiemaatregelen voor de proband kunnen genomen worden en zodat gerichte risicostratificatie bij de relevante familieleden kan gebeuren. In de praktijk wordt slechts in de helft van de gevallen een oorzaak gevonden, waardoor het een plotse onverklaarde dood of SUDS wordt genoemd en er geen gericht familiaal klinisch onderzoek mogelijk is. Hierdoor dreigen belangrijke kansen voor de preventie van familieleden verloren te gaan doordat andere hoogrisico individuen onbekend blijven.
In deze leeftijdsgroep kunnen we de voornaamste etiologie onderverdelen in oorzaken gebaseerd op hereditair structureel hartlijden, een tweede groep erfelijke aandoeningen berustend op primair elektrische hartziekten waarbij er met onze huidige diagnostische technieken doorgaans geen duidelijk structureel hartlijden detecteerbaar is (figuur 1). Minder frequent zijn de thoracale aorta aneurysma en dissecties (TAAD) als derde groep de oorzaak van plotse hartdood.
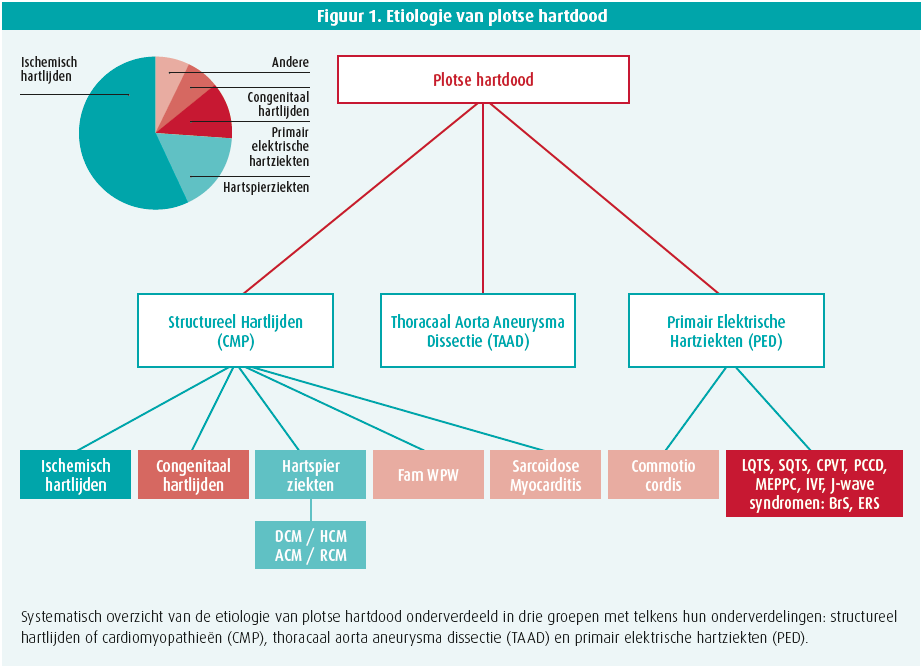
Erfelijke hartspierziekten
De cardiomyopathieën omvatten een heterogene groep van aandoeningen die gekarakteriseerd worden door structurele remodelling die progressief de elektrische stabiliteit van het hart compromitteren. Zo veroorzaakt het primair structureel hartlijden een verhoogd risico tot maligne aritmie en plotse hartdood. De erfelijke cardiomyopathieën die geassocieerd zijn met plotse hartdood kunnen onderverdeeld worden in verschillende groepen: hypertrofe cardiomyopathie (HCM), gedilateerde cardiomyopathie (DCM), aritmogene cardiomyopathie (ACM en restrictieve cardiomyopathie (RCM).
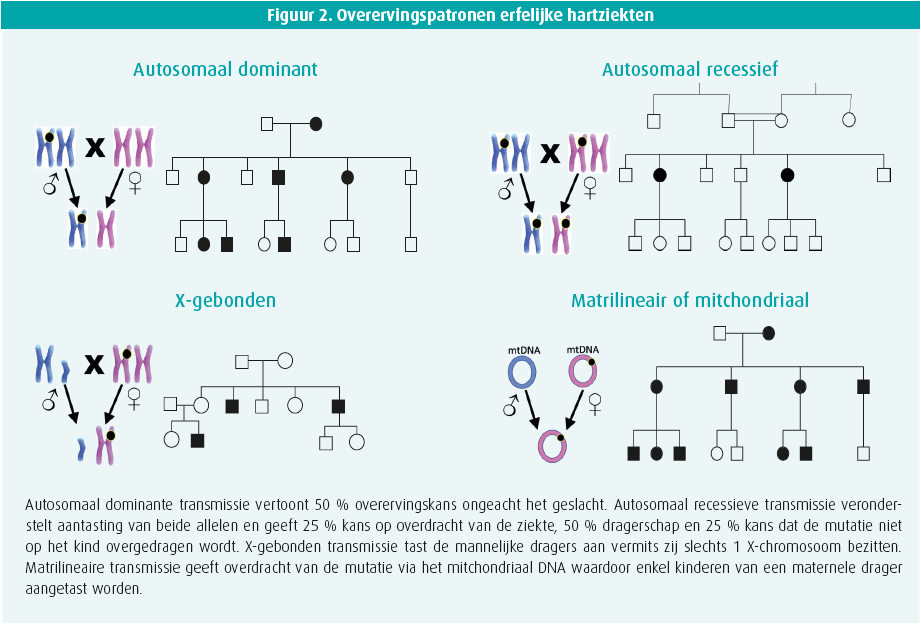
De meeste van deze aandoeningen worden op een autosomaal dominante (AD) wijze overgeërfd (1 allel aangetast, 50 % overervingskans), doch zeldzame autosomaal recessieve (AR) aandoeningen (2 allelen aangetast), X-gebonden en mitochondriale vormen werden beschreven (figuur 2). De aanwezigheid van een genetische variant betekent niet automatisch dat de ziektetekenen of symptomen tot uiting komen. We spreken dan van onvolledige penetrantie. De mechanismen hiertoe zijn niet goed gekend.
Hypertrofe cardiomyopathie (HCM)
HCM wordt gedefinieerd als de asymmetrische excessieve verdikking van het linkerventrikel in de afwezigheid van abnormale belasting. Deze cardiomyopathie is met een prevalentie van 1 per 500 individuen de meest voorkomende erfelijke hartziekte en vertoont een autosomaal dominante overerving. Hierdoor heeft elk eerstegraadsfamilielid van een HCM-patiënt 50 % kans om drager te zijn van dezelfde genetische voorbeschikking tot ontwikkeling van het HCM-fenotype.
De aandoening vertoont een variabele expressieleeftijd en klinische expressie. Het is binnen families met een bewezen mutatie moeilijk voorspelbaar of en in welke mate het fenotype tot uiting zal komen. Het is eveneens bij individuele patiënten niet voorspelbaar of het fenotype zal evolueren naar terminaal systolisch hartfalen, wat in 5-10 % van de HCM-gevallen voorkomt. Plotse hartdood komt voor in 0,4-1 % van de HCM-gevallen, maar subpopulaties met een veel hoger risico werden beschreven. Met de huidige klinische risicostratificatie is het niet mogelijk om het individuele risico nauwkeurig in te schatten.
HCM ontstaat door gendefecten in de sarcomeer-eiwitten (MYBPC3, MYH7, TNNT2, TNNI3 en MYL2), de Z-disc-eiwitten (ACTN2, MYOZ2) en calciumhandling- eiwitten. Een zeldzame vorm van uitgesproken vroege beginnende HCM wordt veroorzaakt door mutaties in het PRKAG2-gen. Dit HCM-fenotype gaat ook gepaard met een familiale vorm van pre-excitatiesyndroom (familiale WPW). Bij kinderen kan HCM in syndromale vorm voorkomen als onderdeel van bv. noonansyndroom, LEOPARD-syndroom, Friedreich's Ataxia (AR), erfelijke metabole en neuromusculaire ziektenv.
Gedilateerde cardiomyopathie (DCM)
Niet-ischemisch gedilateerde cardiomyopathie wordt gekenmerkt door de pathologische uitzetting van de ventriculaire caviteiten en verminderde contractiliteit in de afwezigheid van abnormale belasting. De incidentie wordt geschat op 5-8 per 100 000 en een prevalentie bij volwassenen van 1 per 2 5007.
Hoewel de oorzaak vaak idiopathisch blijft, wordt aangenomen dat de helft van de DCM-gevallen een familiale vorm is. Wanneer er naast een niet-ischemisch gedilateerde cardiomyopathie ook AV-geleidingsstoornissen voorkomen is de kans dat een erfelijke oorzaak aan de basis van het hartlijden ligt groter. Intussen werden meer dan 40 genen geïdentificeerd die betrokken zijn bij de pathogenese van familiale DCM.
Recent werd het titin-gen (TTN) geïdentificeerd als oorzaak van familiale DCM, intussen kan hierdoor ca. 25 % van de idiopathische DCM verklaard worden8. TTN is het grootste in het menselijke genoom voorkomende gen waardoor er ook veel goedaardige varianten teruggevonden worden. De correcte interpretatie van TTN-varianten blijft een grote uitdaging. Tot op heden worden enkel tuncerende (stopcondons en out-of-frame splice site) mutaties gerapporteerd en blijft de beoordeling van puntmutaties tot nog toe het onderwerp van wetenschappelijk onderzoek.
De presentatie van DCM verloopt genspecifiek. Mutaties in het Phospholamban- gen (PLN) veroorzaken een familiale vorm van DCM die op jonge leeftijd evolueert naar systolisch pompfalen9, terwijl mutaties in het Lamin A/C (LMNA/C) gen zijn geassocieerd met een bijzonder maligne vorm van DCM die zich in een vroeg stadium presenteert met bradycardie als gevolg van AV geleidingsstoornissen en maligne aritmie. Evolutie naar systolisch hartfalen treedt meestal later op vanaf de vierde decade. Patiënten met laminopathie hebben doorgaans een slechte prognose in eerste fase door maligne aritmie en later door terminaal hartfalen. Intussen werden genspecifieke risicofactoren voor laminopathie gedefinieerd die toelaten om de preventie van plotse hartdood te verfijnen in deze specifieke populatie10.
X-gebonden DCM komt voor als gevolg van dystrophinemutaties in associatie met musculaire dystrophie (ziekte van Becker en Duchenne), en kan bij mannelijke patiënten bovendien ook solitair voorkomen zonder overte perifere myopathie11.
Artimogene cardiomyopathie (ACM)
Aritmogene cardiomyopathie wordt gekenmerkt door de progressieve epicardiale infiltratie van fibreus en lipomateus weefsel in het myocard. Dit proces leidt tot een progressieve achteruitgang van de systolische functie en tot veranderingen in de normale conductie met elektrische instabiliteit tot gevolg, meestal eerst detecteerbaar ter hoogte van het rechterventrikel. ACM is een ziekte van het adhesie apparaat dat de cardiomyocyten bij elkaar moet houden en tast bij gendefecten meestal beide ventrikels aan, hoewel ook dominant rechter- of linkerventrikelaantasting beschreven werd. De notie dat ACM enkel de rechterventrikel (eerder gekend als ARVC) zou aantasten werd inmiddels verlaten.
De prevalentie van ACM wordt geschat op 1 per 2 000-5 000 individuen. ACM wordt vaak veroorzaakt door desmosomale gendefecten (PKP2, DSP, DSG2, DSC2, JUP) en minder frequent door extra-desmosomale mutaties (oa. RYR2, DES, TMEM43, TGFB3, FLNC en TTN). ACM wordt meestal op autosomaal dominante wijze overgedragen, maar zeldzame autosomaal recessieve vormen zoals de Naxos-ziekte berustend op mutaties in het JUP-gen zijn ook bekend12. Een recente nieuwe groep van ACM met belangrijke uptake zijn de FLNC (Filamine C) mutaties die zowel een aritmogene vorm van hypertrofe als gedilateerde cardiomyopathie kunnen teweegbrengen.13
ACM verloopt in drie fasen met een eerste asymptomatische fase, gevolgd door een aritmogene fase die meestal tot uiting komt op jongvolwassen leeftijd en een derde fase waarbij er ook systolisch hartfalen optreedt. De klinische expressie binnen families is sterk variabel en onvoorspelbaar. ACM is een belangrijke oorzaak voor maligne events en wordt verantwoordelijk geacht voor 30 % van de plotse hartdood bij atleten. Er wordt algemeen aangenomen dat myocarditis betrokken kan zijn bij de pathogenese en dat zware fysieke belasting de progressie van ACM in de hand kan werken. Recent werden ook argumenten voor een oligogenische architectuur aangetoond, wat mogelijk een verklaring is voor de heterogene genotype-fenotype correlatie bij ACM14. De klinische diagnose van ACM is moeilijk en wordt gebaseerd op een conglomeraat van structurele, ECG-grafische, moleculaire en histologische bevindingen die samengebracht onder de Task Force criteria een probabiliteit tot ACM-diagnose opleveren15.
Restrictieve cardiomyopathie (RCM)
RCM is een zeldzame aandoening die gekarakteriseerd wordt door de progressieve verstijving van de hartwand waardoor de diastolische relaxatie belemmerd wordt en dit bij (laag)normale systolische en/of diastolische volumina en in de afwezigheid van significante hypertrofie. Doorgaans evolueert RCM in later stadium naar systolisch hartfalen.
Primaire RCM wordt veroorzaakt door troponinopathie en desminopathie. Troponinopathie wordt gekenmerkt door verhoogd aritmogeen risico terwijl desminopathie zich voornamelijk presenteert aan de hand van AV-geleidingsstoornissen en perifere myopathie.16
Secundaire familiale RCM is het gevolg van myocardinfiltratie zoals bij ATTR-amyloïdose of een gevolg van abnormale stapeling zoals bij hemochromatose en de ziekte van Fabry.
Primair elektrische ziekten
In tegenstelling tot de cardiomyopathieën ontstaan de levensbedreigende hartritmestoornissen bij primair elektrische ziekten zonder de aanwezigheid van detecteerbare structurele hartafwijkingen. Deze groep omvat ziektebeelden zoals: het lange QT-syndroom (LQTS), korte QT-syndroom (SQTS), brugadasyndroom (BS), vroege repolarisatiesyndroom (ERS), catecholaminerge polymorfe ventriculaire tachycardie (CPVT), idiopathische ventrikelfibrillatie (IVF) en progressieve cardiale conductiestoornis (PCCD).
Deze aandoeningen worden veroorzaakt door moleculaire defecten in de ionkanalen die betrokken zijn bij de cardiale actiepotentiaal. Meestal wordt de elektrische instabiliteit veroorzaakt door een functieversterking of functieverlies van een van de cardiale ionstromen waardoor de normale balans tussen depolariserende en repolariserende krachten verstoord wordt. Dit resulteert in een abnormale verlenging of verkorting van de actiepotentiaal, en soms in een abnormale conductievertraging met verhoogde lokale of regionale actiepotentiaal dispersie tot gevolg, een toestand die pro-artimogeen is. Het merendeel van deze aandoeningen wordt veroorzaakt door mutaties in genen die encoderen voor de α-subeenheid van cardiale ionkanalen zoals KCNQ1, KCNH2, SCN5A, CACNA1C, KCNJ2, KCNJ8, KCNJ11, KCND3, TRPM4, SLC4A3 en RYR2. Minder frequent voorkomend zijn mutaties in de β-subeenheden en helper proteïnen van deze ionstromen, bv. CAV3, AKAP-9, SNTA-1, GPD1L, ABCC9, KCNE3, CASQ2, CALM1, CALM2, TRDN, DPP6, MOG1, SLMAP, CACNA2D1, KCNE1, KCNE2, SCNB1 en SCNB3.
Ook hier bestaat dus een belangrijke genetische heterogeniteit. Meerdere genen geven aanleiding tot eenzelfde ziektebeeld wanneer mechanistisch een soortgelijk effect op de actiepotentiaal veroorzaakt wordt. Zo weten we intussen dat wel 16 genen elk een vorm van LQTS kunnen veroorzaken. Anderzijds kunnen mutaties in een gen afhankelijk van de topologie en het type mutatie aanleiding geven tot ziektebeelden die fenotypisch sterk van elkaar verschillen wanneer het mechanistisch effect op het hart verschillend is. Mutaties in het SCN5A-gen kunnen aanleiding geven tot LQTS3, terwijl andere mutaties in dit gen aanleiding geven tot BS, sommige tot gedilateerde cardiomyopathie en weer andere tot Lenegre-Lev's progressieve conductiestoornis (PCCD Type IA).
De functie van cardiale ionkanalen wordt beïnvloed door factoren zoals temperatuur, electrolythuishouding, hormonale balans en PH. Daarnaast bestaat er een complexe farmacogenetische interactie met medicijnen die de uiting van gendefecten kunnen versterken of verzwakken en waarbij de genvariaties die voorkomen in het ionkanaal zelf ook de affiniteit en kinetica van de farmacologische interactie beïnvloeden. Op deze wijze ontstaat er een complexe interactie tussen verworven en erfelijke factoren die allen hun invloed uitoefenen op de polygenisch bepaalde elektrische stabiliteit van het hart. Deze interacties zijn dus allerminst voorspelbaar. Om deze reden is het noodzakelijk om steeds eerst een secundaire oorzaak uit te sluiten wanneer aan primair elektrische ziekten zoals LQTS, SQTS of BS gedacht wordt.
De diagnose van primair elektrische ziekten is vaak moeilijk doordat de diagnostische afwijkingen vaak subtiel en transiënt aanwezig zijn. Ook wordt er binnen families met primair elektrische ziekten een sterk variabele klinische expressie geobserveerd en is de penetrantie vaak beperkt.
Lange QT-syndroom (LQTS)
Het lange QT-syndroom wordt veroorzaakt door de pathologische verlenging van de cardiale actiepotentiaal waardoor de transmurale dispersie toeneemt. De diagnose wordt gesteld na exclusie van secundaire oorzaken aan de hand van recurrente excessieve QTc-verlenging; een toestand die voorbeschikt tot Torsade de Pointes en plotse hartdood. De prevalentie wordt geschat op ca. 1 per 2 500-5 000 individuen. Momenteel zijn er 16 verschillende LQTS-genotypes bekend, 75 % van de erfelijke LQTS-vormen wordt veroorzaakt door mutaties in KCNQ1 (LQST1), KCNH2 (LQST2) en SCN5A (LQTS3). De autosomaal dominante LQTS-vormen werden vroeger het romano-wardsyndroom genoemd. Een zeldzame recessieve vorm die naast LQTS gekenmerkt wordt door congenitale doofheid berust op KCNQ1-mutaties en wordt Jervell Lange-Nielsensyndroom (JLN) genoemd17.
De frequent voorkomende verworven en omkeerbare LQTS-vorm is meestal het gevolg van stoornissen in de electrolythuishouding of het gevolg van farmacologische interactie met QT verlengende medicijnen. Een lijst met QT-verlengende farmaca kan geraadpleegd worden op www.crediblemeds.org. In 10-15 % kan bij de 'verworven' LQTS-vorm toch een onderliggende genetische predispositie aangetoond worden.
Korte QT-syndroom (SQTS)
Het korte QT-syndroom is een zeldzame aandoening die wordt gekenmerkt door excessieve verkorting van de actiepotentiaal, wat zich op het ECG manifesteert aan de hand van QTc-waarden korter dan 330 ms, vaak in combinatie met een verstoorde QT-adaptatie tijdens bradycardie. SQTS veroorzaakt met hoge penetrantie een sterk verhoogd risico voor polymorfe VT/VF met 50 % mortaliteit voor de leeftijd van 40 jaar. Heden worden een 8-tal genen (KCNQ1, KCNH2, KCNJ2, CACNA1C, CACNB2, CACN2D1, SLC22A5 en SLC4A3) verantwoordelijk geacht voor dit syndroom dat autosomaal dominant overgedragen wordt18.
Brugadasyndroom (BS)
Het brugadasyndroom berust op gendefecten die de vroege fase van de actiepotentiaal zodanig beïnvloeden dat lokale epicardiale, meestal transiënte, conductiedefecten ontstaan die proaritmogeen zijn en die voorbeschikken tot VF en plotse hartdood. Plotse hartdood treedt meestal op in rust tussen de 3e en 4e decade en komt vaker voor bij mannen dan bij vrouwen. De diagnose wordt gesteld aan de hand van type-1 ecg-afwijkingen in de rechter precordiale afleidingen en zeldzaam in de inferior afleidingen.
De prevalentie wordt geschat op 1 per 2 000 individuen, hoewel BS in bepaalde gebieden zoals Zuid-Oost Azië veel frequenter voorkomt. BS wordt op autosomaal dominante wijze overgeërfd.
In het verleden werden 23 genen beschreven die geassocieerd waren bij de pathogenese van BS. Recente publicaties tonen aan dat wellicht enkel SCN5A een voldoende sterke klinische impact heeft om op zichzelf het brugadasyndroom te veroorzaken19. Vandaag wordt aangenomen dat de pathogenese van het BS een meer oligogene architectuur vertoont omdat de genotype-fenotypecorrelatie in families meestal niet lineair is en omdat genetische testing slechts 18-32 % van BS-gevallen moleculair kan verklaren. Wellicht spelen de overige genen in sommige families een rol, maar dan eerder in een meer complexe multifactoriële pathogenese. Nieuwe polygene risicoscores worden ontwikkeld om het probleem van de genotype-fenotypecorrelatie en de daaruit voortvloeiende problematische risicostratificatie op te lossen.
Er bestaat ook een verworven medicatie- geïnduceerde vorm van BS. Opnieuw is er een brede waaier aan farmaca die met de ionkanalen kunnen interageren en de ritmestoornissen bij een vooraf bestaand BS bevorderen of een nieuwe omkeerbare vorm van BS (fenocopy) veroorzaken. Het is niet duidelijk of er voor het verworven BS fenocopy ook een onderliggende genetische predispositie aanwezig is. Men kan deze farmaca terugvinden op de website: www.brugadadrugs. org.
Vroeg repolarisatiesyndroom (ERS)
ERS met prominente J-waves op het ECG is meestal een goedaardig fenomeen dat veelvuldig, tot 1-2 % in de algemene populatie en tot 35 % bij normale atleten voorkomt. In zeldzame gevallen kan ≥ 2 mm inferior en/of laterale J-punt elevatie geassocieerd zijn met plotse hartdood. Tot op heden werden 7 genen (KCNJ8, CACNA1C, CACNB2, CACN2D1, ABCC9, SCN5A en SCN10A) geassocieerd met de maligne vorm van ERS.
Catecholaminerge polymorfe ventriculaire tachycardie (CPVT)
CPVT is een zeldzame aandoening die gekenmerkt wordt door het optreden van polymorfe ventrikeltachycardie of bidirectionele tachycardie in toestanden van verhoogde adrenerge tonus, bv. inspanning en hevige emotie. De ritmestoornissen ontstaan door een intracellulaire calciumoverbelasting in de cardiomyocyt die aanleiding geeft tot late nadepolarisaties die op hun beurt een maligne ritmestoornis kunnen uitlokken.
De prevalentie wordt geschat op 1 per 10 000 individuen. Het rust-ecg vertoont geen kenmerkende afwijkingen. Echter, wanneer de hartfrequentie toeneemt boven 100-120 spm ontstaan meer en meer multifocale PVC's die progressief meer polymorf en sustained worden wanneer de hartfrequentie verder accelereert. In vele gevallen ontstaat uiteindelijk een bidirectionele ventrikel tachycardie die gekarakteriseerd wordt door de 180° asrotatie die na elk ventriculair complex optreedt. Deze aritmie kan degenereren in VF en plotse hartdood.
Momenteel zijn er 6 oorzakelijke genen voor CPVT bekend (RYR2, CASQ2, TRDN, CALM1, CALM2, CALM3) die de moleculaire verklaring vormen van ongeveer 60 % van de CPVT gevallen20. CPVT wordt autosomaal dominant overgedragen, behalve CPVT berustend op gendefecten in CASQ2 en TRDN die een autosomaal recessieve transmissie vertonen.
De penetrantie van het CPVT-fenotype (syncope, maligne aritmie en/of plotse hartdood) wordt geschat op 80 % voor de leeftijd van 40 jaar. Het is dan ook belangrijk om steeds alle asymptomatische patiënten te identificeren en te behandelen om events te voorkomen.
Idiopathische ventrikelfibrillatie (IVF)
IVF is een verzamelnaam voor aandoeningen die plotse hartdood veroorzaken door ventrikelfibrillatie zonder aantoonbare omkeerbare oorzaak of onderliggend structureel of elektrisch hartlijden. Het basis-ecg vertoont meestal geen afwijkingen en VF wordt doorgaans uitgelokt door monofocale kort gekoppelde PVC's. De monofocale origine van deze vaak purkinje-gerelateerde PVC's maakt dat ablatie de aritmie burden significant kan reduceren. Het is dus van belang patiënten met plotse hartdood langdurig te monitoren om dit soort kort gekoppelde PVC's die vaak aanleiding geven tot korte runs meer complexe ritmestoornissen te detecteren. Bij familiale IVF vormen werden mutaties teruggevonden in SCN5A en DPP621.
Cascade-onderzoek
Het doel van cardiogenetica en plotse hartdood management is de optimale behandeling van de onderliggende aandoening en de preventie van recidief bij de proband. Daarnaast willen we hoogrisico familieleden identificeren om indien nodig preventiemaatregelen en/of de gepaste behandeling op te starten.
Het cardiogenetisch onderzoek start steeds bij de proband die verondersteld wordt de meest uitgesproken expressie te vertonen van de onderliggende oorzakelijke ziekte. De genotype-fenotypecorrelatie wordt dus verondersteld het duidelijkste te zijn bij de proband. Alvorens het genotype in kaart te brengen is het dus van cruciaal belang om het fenotype bij de proband zo volledig mogelijk te karakteriseren. Het zal dienen als leidraad voor verdere risicostratificatie en helpen bij de interpretatie van de genetische gegevens. Op basis van het bekomen genotype-fenotype profiel kunnen vervolgens de eerstegraadfamilieleden onderzocht worden, wat dan kan leiden tot onderzoek bij de tweedegraadfamilieleden, en zo verder.
Elk cardiogenetisch onderzoek moet dus in de eerste plaats gedreven worden door de aanwezigheid van een fenotype. Naast risicostratificatie kan het cardiogenetisch onderzoek bij de familieleden ook helpen om het familiale fenotype en de klinische heterogeniteit beter in kaart te brengen.
Gezien de klinische heterogeniteit, onvolledige penetrantie en onzekerheden over symptomen en prognose die soms blijven bestaan bij cardiogenetische aandoeningen moet de beslissing tot verder familiaal onderzoek steeds kritisch afgewogen worden tegenover de mogelijke schade die aangericht kan worden door de impact op het leven, angst en psychologische draagkracht22. Dit wordt uiterst belangrijk wanneer het onderzoek ook van toepassing is op jonge kinderen. Deze complexe afweging moet dan ook steeds in multidisciplinaire zorgteams worden gemaakt, meestal bestaande uit een klinisch cardioloog met oriëntatie in erfelijke cardiale ziekten, klinische geneticus, moleculaire geneticus, genetische counselor, psycholoog en een cardiogenetisch verpleegkundige.
Fenotypering
Het cardiovasculair onderzoek moet ten minste een gedetailleerde persoonlijke en familiale anamnese, klinisch onderzoek, rust-ecg, inspannings-ecg, echocardiografie en 24u-holter met QT/QTc monitoring omvatten. Dit kan indien nodig aangevuld worden met ziektespecifieke onderzoeken zoals oa. CMR, cardiale CT, ajmalinetest, SAECG, inspanningsechocardiografie en EFO. Vervolgens wordt een stamboom opgesteld om het familiale fenotype beter in kaart te brengen en om andere informatieve familieleden te identificeren.
Tijdens de anamnese moet speciale aandacht besteed worden aan alarmsymptomen en dient een grondige bevraging van doorgemaakte maligne events te gebeuren aangezien hierdoor belangrijke aanwijzingen over de oorzaak kunnen bekomen worden.
Alarmsymptomen
Het symptoom dat bij uitstek oriënteert naar een maligne hartritmestoornis is de cardiogene syncope. Dit is een syncopaal event dat zeer bruusk optreedt zonder prodromen en met volledig kortstondig bewustzijnsverlies tot gevolg. Doorgaans treedt er na enkele seconden volledig herstel van het bewustzijn op. Er is geen post-ictale fase. Het voorkomen van incontinentie is een teken van spieratonie als gevolg van hypotensie en draagt niet bij tot differentiële diagnose van het event. Een bruuske syncope is een alarmsymptoom dat altijd bijkomend cardiaal onderzoek vereist.
Syncopes die optreden tijdens (niet na) inspanning of in liggende houding zijn ook steeds verdacht voor een cardiaal event. Het is belangrijk dit actief te bevragen tijdens de anamnese, aangezien patiënten plots bewustzijnsverlies niet steeds associëren met cardiale problemen. Vaak wordt dit omschreven als een 'appelflauwte' of 'black-out' en verder genegeerd.
Symptomatische palpitaties met presyncope is een aanwijzing voor een hartritmestoornis met een significante hemodynamische impact. Dit kan het gevolg zijn van zowel supraventriculaire als ventriculaire ritmestoornissen en verdient steeds gericht bijkomend onderzoek.
Early onset voorkamerfibrillatie, angina pectoris en hartfalen kunnen een marker zijn van respectievelijk verhoogde elektrische instabiliteit zoals gezien wordt bij BS en LQTS, relatieve ischemie bij hypertrofe cardiomyopathie en hartfalen als gevolg van een onderliggende cardiomyopathie. Patiënten met onverklaarde epilepsie die met EEG nooit gedocumenteerd werd, blijkt soms een epileptiforme syncope als gevolg van maligne aritmie te zijn. Absences bij kinderen kunnen soms het gevolg zijn van abnormale bradycardie zoals gezien kan worden bij BS.
Hyperventilatie gaat meestal gepaard met angst, subjectieve distress en hartkloppingen. In sommige gevallen wordt de hyperventilatie-aanval uitgelokt door symptomatische palpitaties.
Plotse dood
Bij plotse hartdood is het belangrijk om na te gaan in welke omstandigheden het event plaatsvond. Indien mogelijk worden de getuigen gecontacteerd aangezien zij vaak waardevolle informatie kunnen verschaffen over het verloop en de omstandigheden van het event.
Vervolgens is het van groot belang om het eerste gedocumenteerde ritme op te vragen bij de urgentiediensten die de reanimatie uitvoerden. Ook wanneer een AED gebruikt werd, kan een uitlezing van het event opgevraagd worden. De documentatie van een Torsade de Pointes of bidirectionele VT is vrijwel pathognomonisch voor respectievelijk LQTS en CPVT.
Bij recurrente aritmie kunnen de basisfrequentie en het initiatiepatroon en koppelingsinterval (bv. kort gekoppelde PVC vs. pauze-afhankelijke initiatie, kort-lang-kort sequentie, progressieve degeneratie van PVC naar pVT/VF) bijkomende aanwijzingen opleveren over de oorzaak. Eventuele monitoring devices zoals ILR, pacemaker of ICD worden daarom best uitgelezen.
Autopsie of post-mortem fenotypering
Wanneer de patiënt overleden is, wordt het belangrijk een autopsie te laten plaatsvinden. Dit kan belangrijke aanwijzingen over onderliggend structureel hartlijden (of de totale afwezigheid ervan) aan het licht brengen, toxicologische oorzaken uit te sluiten en laat toe om een DNA-staal te bekomen voor verder genetisch onderzoek. Wanneer de patiënt overlijdt buiten het ziekenhuis kan enkel een DNA-staal afgenomen worden voordat het overlijdensattest ingevuld werd. Wel kunnen urgentieteams getraind worden om de endotracheale tube of het intra-osseeuze access als spijtstaal te bewaren om te versturen naar het cardiogenetisch centrum voor DNA-extractie en -analyse.
Ook hier kunnen ritmemonitoring devices uitgelezen worden om de artimogene oorzaak van de plotse hartdood te achterhalen. In de praktijk gebeurt dit alles te weinig waardoor de enige mogelijkheid tot structurele informatie en genetisch onderzoek vaak verloren gaat. Hierdoor ontstaat een situatie waarin elke fenotypische informatie ontbreekt.
Het SUDS-fenotype
Wanneer de patiënt overleden is als gevolg van het event en er geen aantoonbare oorzaak kan worden gedetecteerd, spreken we van plotse dood van ongekende oorzaak of SUDS. Vaak werd er geen autopsie uitgevoerd of kon de obductie geen doodsoorzaak verklaren. Dit bijzondere fenotype bemoeilijkt de mogelijkheden tot familiale preventie tenzij tijdens de autopsie een weefselstaal of bloedstaal afgenomen werd. In dat geval kan alsnog een moleculaire autopsie uitgevoerd worden die de diagnose aan het licht kan brengen en ons in staat kan stellen andere hoogrisico familieleden te identificeren en te beschermen.
Genotypering
Na grondige fenotypering bij de proband wordt een EDTA-bloedstaal afgenomen voor genetische analyse. Bij autopsie wordt een weefselstaal, meestal leverweefsel, verstuurd voor genetisch onderzoek.
Diagnose
Het genetisch onderzoek heeft tot doel de diagnose bij de proband te bevestigen wanneer er een duidelijk fenotype aanwezig is. Wanneer het fenotype onduidelijk is, of wanneer er helemaal geen fenotype bekend is kan het diagnostisch cardiogenetisch onderzoek helpen om de diagnose vast te stellen.
Ook in het geval van overlijden door SUDS (zonder aanwijsbare oorzaak) en wanneer autopsie niet werd uitgevoerd werd of negatief was, kan de post-hoc genotypering of moleculaire autopsie helpen om alsnog de diagnose te stellen. Deze gespecialiseerde techniek laat toe om de diagnose in ca. 25 % van de onopgeloste jonge plotse overlijdens te stellen waardoor hoogrisico familieleden toch beter beschermd kunnen worden.
Eens een ziekmakende mutatie geïdentificeerd wordt, kan eenvoudig met zekerheid en onafhankelijk van de aanwezigheid van ziektetekenen vastgesteld worden of een familielid drager is van dezelfde ziekmakende factor. Dit predictief genetisch onderzoek laat dus toe om in een presymptomatisch stadium te voorspellen of het fenotype bij de persoon tot uiting kan komen. Op deze wijze maakt het cardiogenetisch onderzoek het mogelijk om patiënten in een presymptomatisch stadium op te volgen, de nodige primaire preventieve maatregelen te nemen om ziekte manifestatie te vertragen/voorkomen, en desgevallend in een vroegtijdig stadium de ziektetekenen en of vroege (alarm)symptomen te detecteren.
Bovendien is het mogelijk om te voorkomen dat een ziekmakende mutatie met potentieel belangrijke fenotypisch impact wordt overgedragen naar een volgende toekomstige generatie. Via invitrofertilisatie geeft pre-implantatie genetische testing (PGT) waarbij het dragerschapsonderzoek gebeurt op één cel van een vroegstadig embryo, de mogelijkheid om enkel het embryo dat geen drager is van de ziekmakende mutatie terug te plaatsen. Zo voorkomt men transmissie en kan de ziekte familiaal gestopt worden. In sommige publicaties wordt dit wel eens primordiale preventie genoemd.
Tenslotte stelt genotypering ons in staat om ziektebeelden beter van elkaar te differentiëren. Zo zal de groep patiënten die nu nog onder de werkdiagnose 'ideopathisch gedilateerde cardiomyopathie' verzameld worden, geleidelijk aan verkleinen. Nu weten we inmiddels dat er ook oa. aritmogene cardiomyopathieën, laminopathieën, desminopathieën, phospholamban cardiomyopathie, titin cardiomyopathie bestaan met elk hun natuurlijke ziekteverloop en met specifieke noden tot preventie en behandeling. Om deze laatste reden zullen we voor een aantal van deze aandoeningen evolueren naar genspecifieke moleculaire diagnose, zoals voor LMNA/C, DES en PLN al het geval is.
Preventie
Naast opties tot primaire en primordiale preventie faciliteert het cardiogenetisch onderzoek in sommige gevallen de beslissing tot onmiddellijke secundaire preventiemaatregelen. Niet zelden worden alarmsymptomen zoals cardiogene syncopes niet als dusdanig herkend en worden vaak door patiënten genegeerd. Wanneer blijkt dat zulke alarmsymptoom in combinatie met de cardiogenetische diagnose een sterk verhoogd risico voor plotse hartdood voorstelt, kan een ICD noodzakelijk zijn om de patiënt te behoeden voor plotse hartdood.
Door de snelle vooruitgang in onze cardiogenetische technieken en de aanleg van wereldwijde registers en databanken wordt het ook meer en meer duidelijk dat genotypering ook rechtstreeks kan bijdragen tot de risicostratificatie van de proband en zijn familieleden. Op genniveau en in combinatie met andere risicofactoren weten we dat frameshift- en truncatiemutaties in het LMNA/C-gen een hoger plotsedoodrisico met zich dragen dan puntmutaties, wat bij mannelijke dragers voldoende reden kan zijn om ICD-implantatie te overwegen10.
In zeldzame gevallen is het genotype in combinatie met de familiale expressie al voldoende, zoals bv. bij atypische CPVT waar de aangetaste patiënten met geen enkele klinische test een aantoonbaar fenotype hebben, gaat dragerschap van de familiale RYR2-mutatie gepaard met een extreem hoge incidentie voor plotse doodevents in de familie waardoor dragerschap van de pathogene mutatie op zich de implantatie van een ICD kan verantwoorden.
Intussen werd ook aangetoond dat sommige specifieke mutaties een uitermate hoog risico met zich meebrengen zoals bv. p.Arg92Gln (of p.Arg102Gln) in TNNT2 die een maligne vorm van HCM veroorzaakt gekenmerkt door een mortaliteit van 50 % voor de leeftijd van 50 jaar23. Het spreekt voor zich dat hoewel onze klassieke risicostratificatie voor HCM dit niet in rekening zou brengen en bij gevolg onze terugbetalingscriteria niet aangepast zijn, dragerschap van dit genotype op zich voldoende moet zijn om een ICD-implantatie ter preventie van plotse hartdood te verantwoorden. Zo werd door samenwerking tussen de cardiogenetische centra recent ook een Belgische founder mutatie geïdentificeerd in het SCN5A-gen die een meer maligne vorm van BS veroorzaakt24.
Behandeling
Genotypering kan ook een rechtstreekse impact hebben op het management van patiënten. Op gen-niveau weten we dat een type-1 LQTS gebaseerd op KCNQ1-mutaties anders behandeld zal worden dan LQTS2 berustend op KCNH2-mutaties en anders dan LQTS3 als gevolg van SCN5A-mutaties aangezien de ionkanalen van elk van deze genen een andere specifieke functionele expressie kent in de cascade van de cardiale actiepotentiaal. Zo komen ritmestoornissen bij LQTS1 meer voor bij snelle hartfrequenties terwijl dit bij LQTS2 minder en bij LQTS3 eerder bij trage frequenties gebeurt.
Intussen is de cardiogenetica ook geëvolueerd tot mutatiespecifieke therapie. 'Gain-of-function' mutaties in het SCN5A-gen zoals de p.Arg222Gln-mutatie die de functie van het natriumkanaal excessief versterkt, kunnen aanleiding geven tot hyperexciteerbaarheid met zeer frequente extrasystolie tot gevolg. Vaak geeft dit op jonge leeftijd aanleiding tot een vorm van tachycardiomyopathie met hartfalen tot gevolg. Deze nieuwe entiteit wordt 'Multifocale Ectopische Purkinje- afhankelijke Premature Contracties' of kortweg MEPPC genoemd25. Uit eigen ervaring weten we dat ablatietherapie van deze extrasystolen onmogelijk is door het extreme multifocale aspect als teken van het hypergeëxciteerd myocard. Het is vanwege het gevaar voor pro-aritmie tegenaangewezen om Vaughan Williams klasse Ic natriumkanaalblokkers zoals flecaïnide te gebruiken bij patiënten met verminderde LVEF. Toch is hier een duidelijke translatie van een bewezen 'gain-of-function' natriumkanaal genotype naar het hyperexciteerbaarheid fenotype met secundaire tachycardiomyopathie. Behandeling met een natriumkanaalblokker grijpt rechtstreeks in op dit ziekmakende mechanisme, reduceert de genetisch versterkte natriumkanaalfunctie, doet de hyperexciteerbaarheid verdwijnen en leidt bij deze patiënten doorgaans binnen de drie maanden tot volledige restitutie van de LV-functie en verdwijnen alle klachten. Wederom blijkt dat genotypering de behandeling voor specifieke patiënten kan verfijnen en dat oude paradigma's wellicht te algemeen zijn voor de translationeel moleculaire cardiologie van de nabije toekomst.
Wat is een ziekmakende mutatie?
Het menselijke genoom bevat grofweg 21 000 genen. In elk van deze genen kunnen variaties optreden. Jarenlang dacht men dat een genetische variatie in een gen ook automatisch de oorzaak was van de ziekte waarvoor men een genetische oorzaak trachtte aan te tonen. Nu weten we dat genetische afwijkingen kunnen behoren tot normale evolutieve variatie en niet steeds ziekmakend zijn. In regel worden in de populatie frequent voorkomende genetische varianten, polymorfismen genoemd, als goedaardig beschouwd. Het voorkomen van de genetische varianten in een controlepopulatie wordt de 'minor allel frequentie' (MAF) genoemd. Het frequent voorkomen van een variant in de algemene populatie (hoge MAF > 1 %) wordt beschouwd als een teken dat deze variant in de evolutie beter getolereerd werd en dus wellicht geen belangrijke ziekmakende impact heeft gehad waardoor deze zich veelvuldig heeft kunnen ontwikkelen en verspreiden. Omgekeerd wordt de kans op pathogeniciteit van zeldzame genetische variatie hoger ingeschat omdat deze evolutief minder kans hebben om doorgegeven te worden, zeker wanneer het fenotype uitmondt in een hoger risico op plotse hartdood.
Er bestaan echter zeer frequent voorkomende polymorfismen die op zich geen ziekte veroorzaken, maar die in combinatie met andere genetische varianten het fenotype kunnen amplificeren of verzwakken. Dit soort varianten die slechts in een specifieke constellatie van genetische variatie klinisch relevant worden, noemen we 'disease modifiers'.
Zeker voor de elektrische hartziekten is dit belangrijk, aangezien de cardiale actiepotentiaal gevormd wordt door een complexe cascade van ionstromen die elkaar beïnvloeden. Een kleine verandering in de functionele expressie van een component kan gevolgen hebben op een of meer andere componenten. Zo werd functioneel aangetoond dat twee varianten, p.Lys897Thr en p.Ala1116Val, in het KCNH2-gen dat encodeert voor het IKr kaliumkanaal en die op zich geen ziekmakend potentieel hebben, wanneer ze samen voorkomen wel degelijk LQTS teweegbrengen. p.Lys897Thr in KCNH2 komt voor in 27 % van de Kaukasische populatie. Zogenaamde polymorfismen kunnen dus wel degelijk ziektemodifiërende eigenschappen vertonen26. Uiteraard is ook niet elke zeldzame variant noodzakelijk betrokken bij het ziekteproces.
Het wordt meteen ook duidelijk dat genetische ziekten niet altijd een monogenische architectuur vertonen, aangezien vaak meerdere genetische varianten in verscheidene genen een rol spelen bij het ontstaan van de uiteindelijke genotype- fenotype-expressie.
We leren hieruit dat een variant in een patiënt ziekmakende eigenschappen kan vertonen terwijl dezelfde variant in een ander individu of familielid afhankelijk van de aanwezigheid van andere schijnbaar banale genetische varianten, helemaal geen fenotype zal teweegbrengen. Het geheel van genetische factoren die nog een rol kunnen spelen bij het ontstaan van de genotype-fenotypecorrelatie noemen we de genetische achtergrond en die is voor elk individu, ook binnen families, sterk verschillend. Aan de andere kant kennen we ziekmakende mutaties die een milder ziekte fenotype veroorzaken dat bovendien slechts op late leeftijd, ver na de reproductieve leeftijd, tot uiting komt waardoor het evolutief selectiemechanisme niet gewerkt heeft en waardoor deze mutaties ongestoord segregeren over de generaties heen. Zulke mutaties vertonen dan ook een ongewoon hoge frequentie in de populatie27.
Zo ontstaat er dus een spectrum van pathogeniciteit dat varieert van banale varianten, naar polymorfismen, 'disease modifiers' tot pathogene mutaties. De genetische achtergrond van elke persoon is verschillend, waardoor de inschatting van de pathogeniciteit van genetische varianten steeds individueel en met uiterste voorzichtigheid moet gebeuren.
Classificatie van genetische varianten
Momenteel worden de genetische varianten naar pathogeniciteit ingedeeld in 5 klassen; benigne (UVKL1), waarschijnlijk benigne (UVKL2), ongekende klinische betekenis (UVKL3), waarschijnlijk pathogeen (UVKL4) en pathogeen (UVKL5). De variant classificatie gebeurt volgens de ACMG-richtlijnen28 en houdt rekening met het voorkomen van de variant in controlepopulaties, ziekte-specifieke en genetische sequentiedatabanken zoals LOVD, GnomAD en ExAC. Daarnaast is het type genetische variatie belangrijk, zoals bv. truncatiemutaties, die grote veranderingen teweegbrengen in het resulterende eiwit bezitten een groter ziekmakend potentieel dan puntmutaties. Verder wordt de pathogeniciteit van genetische varianten ingeschat a.d.h.v. de filogenetische conservering. Aminozuurposities die over de species heen sterk geconserveerd zijn blijken evolutief erg belangrijk te zijn. Varianten op deze posities worden dan ook meer pathogeen ingeschat. Ten slotte wordt op basis van 'in silico' predictie algoritmen de impact op de primaire, secundaire en tertiaire eiwitstructuur ingeschat als maat voor pathogeniciteit.
Enkel voor waarschijnlijk pathogene (UVKL4) en pathogene (UVKL5) varianten is er voldoende correlatie tussen genotype en fenotype aangetoond, waardoor enkel deze varianten kunnen gebruikt worden voor verder predictief cardiogenetisch onderzoek bij familieleden.
Benigne (UVKL1) en waarschijnlijk benigne (UVKL2) varianten hebben geen klinische betekenis en worden om deze reden niet gerapporteerd.
Veruit de grootste groep varianten die heden gevonden wordt, zijn de varianten van ongekende klinische betekenis (UVKL3) waarvoor er aanwijzingen zijn dat de variant betrokken kan zijn bij de etiologie van het fenotype maar onvoldoende om causaliteit te bewijzen of te verwerpen. Klasse 3-varianten kunnen om deze reden niet gebruikt worden voor predictieve genetische testing van presymptomatische familieleden. In sommige gevallen kan co-segregatie analyse de diagnostische betekenis van klasse 3-varianten verder uitklaren waardoor herclassificatie naar informatieve pathogeniciteitsklassen 4 of 5 mogelijk wordt.
Indicaties voor cardiogenetisch onderzoek
Multidisciplinair team
Vermits cardiogenetische ziekten bij leven en overlijden een aanpak vergen waarvoor een brede kennis noodzakelijk is, wordt het cardiogenetisch onderzoek best gevoerd door multidisciplinaire teams die idealiter bestaan uit cardiologen met specifieke training in volwassen en pediatrische cardiogenetische ziekten, moleculaire en klinische genetici en pathologen met specialisatie in hartaandoeningen. Het team wordt best ondersteund door genetische counselors, psychologen en verpleegkundigen. Zo kan een gespecialiseerde doch brede holistische aanpak gegarandeerd worden.
Genetische counseling
Genetische counseling wordt sterk aanbevolen voor alle families waar een plotse hartdood event is voorgekomen en waarbij een erfelijke oorzaak vermoed wordt. Ook wanneer de patiënt overleden is en de oorzaak niet kan vastgesteld worden, is genetische counseling noodzakelijk.
Dit laat toe de nodige informatie over ziektegeschiedenis, alarmsymptomen, verloop van het event van de proband te verzamelen, een stamboom van de familie op te stellen en een hypothese over de mogelijk onderliggende oorzaak voorop te stellen. Aldus worden het fenotype en eventuele familiale fenotype in kaart gebracht zodat een gewogen inschatting kan gemaakt worden over het nut, de voor-en nadelen, en de te verwachten resultaten van een genetisch onderzoek.
Hierbij blijft steeds belangrijk om de mogelijkheid tot psychologische ondersteuning aan te bieden voor de overlevende proband en zijn familieleden.
Diagnostisch onderzoek
Wanneer een oorzakelijke erfelijke hartziekte wordt vermoed bij plotse hartdood bestaat er een harde indicatie tot genetisch onderzoek om de moleculaire oorzaak te achterhalen en om de identificatie van (pre-symptomatische) hoogrisico familieleden met dragerschapsonderzoek via cascadescreening toe te laten. In eerstegraad familieleden van een proband met plotse hartdood met vermoedelijke erfelijke hartziekte wordt cardiale fenotypering aanbevolen en waar mogelijk ook genetische cascade screening toegepast29.
Bij plotse onverklaarde hartdood en wanneer de oorzaak na expert multidisciplinaire evaluatie onverklaard blijft, maar toch met aanwijzingen voor een aritmogene oorzaak is genetisch onderzoek met de focus op onderliggende primair elektrische hartziekten aanbevolen. Bij jonge onverklaarde SUDS kan genetisch onderzoek naar elektrische hartziekten overwogen worden.
Voor de eerstegraad familieleden is het aanbevolen om het cardiale screeningonderzoek elke 3-5 jaren te herhalen en eerder wanneer meer dan één plotse hartdood in de familie is voorgekomen.
Predictief onderzoek
Het (pre)symptomatisch dragerschapsonderzoek laat toe familieleden met een voorbeschikking tot het familiale fenotype te identificeren. Het predictief genetisch onderzoek wordt voorbehouden voor klasse 4 en 5 waarschijnlijk pathogene en bewezen pathogene varianten.
Pre-implantatie genetische testing (PGT)
De mogelijkheid om transmissie van ziekmakende genetische mutaties te voorkomen wordt voorbehouden voor klasse 4 waarschijnlijk pathogene en klasse 5 pathogene varianten. Hiertoe wordt een IVF-traject opgestart met single-cell dragerschapsonderzoek gevolgd door embryoselectie.
Besluit
Cardiogenetica is de sleutel tot een moleculaire dimensie van cardiologie die onze mogelijkheden tot risicostratificatie, preventie en behandeling nog verder kan verbeteren en individualiseren. Hoewel we nog maar aan het begin staan van deze nieuwe sprong in onze kennis en begrip over hartziekten is dit wel al onze nieuwe realiteit. Elk van de hoger vernoemde mutaties werd in de Belgische populatie aangetroffen. Ondanks de snelle vooruitgang blijken vele varianten nog te weinig informatief om klinisch bruikbaar te zijn. Wellicht zal artificiële intelligentie voor betere fenotypering en big data-analyse voor betere variantanalyse ons helpen om de specifieke betekenis van varianten voor de individuele patiënt in polygenische achtergrond te interpreteren.
De voorwaarden om dit alles mogelijk te maken, blijven de afname van een DNA-staal bij elk overlijden met verdenking van cardiogenetische oorzaak, de correcte genotype-fenotypecorrelatie en de doorgedreven multidisciplinaire aanpak in gespecialiseerde cardiogenetische centra op weg naar een betere preventie van plotse hartdood.
Referenties
- Hasselqvist-Ax, I. et al. Early cardiopulmonary resuscitation in out-ofhospital cardiac arrest. N Engl J Med, 2015, 372 (24), 2307-2315.
- Perkins, G.D. et al. European Resuscitation Council Guidelines 2021: Executive summary. Resuscitation, 2021, 161 (1).
- O'Mahoney, C. et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J, 2014, 35 (30), 2010-2020.
- Van der Werf, C. et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in The Netherlands. Heart Rhythm, 2010, 7 (10), 1383-1389.
- Hendrix, A. Sudden cardiac death in the young: incidence and consequences. University of Utrecht, 2011.
- Sabater-Molina, M. et al. Genetics of hypertrophic cardiomyopathy: A review of current state. Clin Genet, 2018, 93 (1), 3-14.
- Jefferies, J.L. et al. Dilated cardiomyopathy. The Lancet, 2010, 375 (9716), 752-762.
- Herman, D.S. et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med, 2012, 366 (7), 619-628.
- Schmitt, J.P. et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science, 2003, 299 (5611), 1410-1413.
- van Rijsingen, I.A. et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol, 2012, 59 (5), 493- 500.
- Arbustini, E. et al. Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J Am Coll Cardiol, 2000, 35 (7), 1760-1768.
- Elliott, P.M. et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail, 2019, 21 (8), 955-964.
- Corrado, D. et al. Filamin C: A New Arrhythmogenic Cardiomyopathy-Causing Gene? JACC Clin Electrophysiol, 2018, 4 (4),515-517.
- Ye, J.Z. et al. Reevaluation of genetic variants previously associated with arrhythmogenic right ventricular cardiomyopathy integrating population-based cohorts and proteomics data. Clin Genet, 2019, 96 (6), 506-514.
- Markus FI et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J, 2010, 31 (7), 806-814.
- Arbustini, E. et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail, 2006, 8 (5), 477-483.
- Shimizu, W. et al. Genetics of long-QT syndrome. J Hum Genet, 2016, 61 (1), 51-5.
- Raschwitz, L.S. et al. Differences in Short QT Syndrome Subtypes: A Systematic Literature Review and Pooled Analysis. Front Genet, 2020, 10, 1312.
- Hosseini, S.M. et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation, 2018, 138 (12), 1195-1205.
- Wleklinski, M.J. et al. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J Physiol, 2020, 598 (14), 2817-2834.
- Postema, P.G. et al. Founder mutations in the Netherlands: familial idiopathic ventricular fibrillation and DPP6. Neth Heart J, 2011, 19 (6), 290-296.
- Manrai, A.K. et al. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med, 2016, 375 (7), 655-665.
- Ripoll-Vera, T. et al. Clinical and Prognostic Profiles of Cardiomyopathies Caused by Mutations in the Troponin T Gene. Rev Esp Cardiol (Engl Ed), 2016, 69 (2), 149-158.
- Sieliwonczyk, E. et al. Clinical characterization of the first Belgian SCN5A founder mutation cohort. Europace, 2020, euaa305.
- Laurent, G. et al. Multifocal ectopic Purkinjerelated premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol, 2012, 60 (2), 144-156.
- Crotti, L. et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation, 2005, 112 (9), 1251-1258.
- Buxbaum, J.N. et al. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med, 2017, 19 (7), 733-742.
- Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17 (5), 405-424.
- Stiles, M et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm, 2021, 18 (1), e1-e50.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.