Compte rendu du congrès de l'EHRA
Introduction
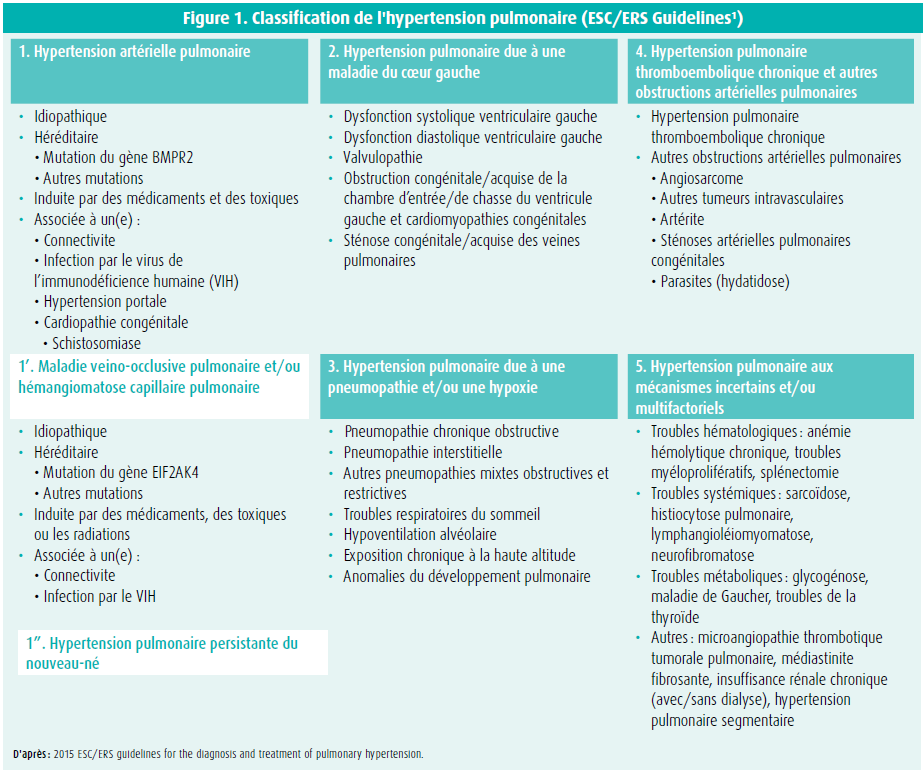
L'hypertension pulmonaire (HTP) est une condition physiopathologique définie par une augmentation de la pression artérielle pulmonaire moyenne au-delà de 25 mmHg, mesurée au repos par cathétérisme cardiaque droit. La classification clinique de l'HTP permet de distinguer cinq groupes différents, associant des pathologies qui présentent des caractéristiques communes en termes de présentation clinique, de mécanismes physiopathologiques, d'hémodynamique et de stratégie thérapeutique. Les formes d'HTP les plus fréquentes sont le groupe 2 (l'hypertension pulmonaire liée aux affections cardiaques gauches) et le groupe 3 (l'hypertension pulmonaire lié aux maladies respiratoires et/ou à l'hypoxémie). L'hypertension artérielle pulmonaire (HTAP) (groupe 1) est une maladie rare, grave, rapidement progressive et incurable. Elle est soit idiopathique, héréditaire, liée à une exposition à des toxiques, associée à des connectivites, à l'infection par le VIH, à l'hypertension portale ou à des cardiopathies congénitales. Il est donc nécessaire de distinguer ces différentes entités, car certaines sont associées à des prises en charges thérapeutiques spécifiques, ce qui est notamment le cas de l'HTAP et de l'hypertension pulmonaire liée à une maladie thrombo-embolique chronique (CTEPH - groupe 4) (figure 1)1.
Définition hémodynamique
La définition hémodynamique de l'HTP (PAPm ≥ 25 mmHg) tient compte de la valeur de pression artérielle pulmonaire occluse (PAPO), qui lorsqu'elle est inférieure ou égale à 15 mmHg permet de définir une hypertension pulmonaire pré-capillaire et regroupe les HTP du groupe 1, 3, 4 et 5. Lorsque la PAPO est > 15 mmHg, on parlera d'hypertension pulmonaire post-capillaire (groupe 2 et 5), qui sera soit isolée (IpcPH), soit combinée (CpcPH). L'hypertension pulmonaire post-capillaire isolée est définie par un gradient diastolique pulmonaire (GDP = PAPd-PAPO) < 7 mmHg et/ou une résistance vasculaire pulmonaire (RVP) ≤ 3 UW. L'hypertension pulmonaire post-capillaire combinée est définie par un GDP ≥7 mmHg et/ou une RVP > 3 UW1. Comme cité précédemment, seul le cathétérisme cardiaque droit permet de confirmer le diagnostic d'HTP et d'en caractériser la présentation. Quels seront donc les patients à considérer comme suspects d'HTP et qui devront in fine être référés vers des centres d'expertise pour réaliser un cathétérisme cardiaque droit ?
Démarche diagnostique
La démarche à suivre pour poser le diagnostic d'HTP est représentée dans cet algorithme décisionnel publié par l'ESC dans les « Guidelines for diagnosis and treatment of pulmonary hypertension » de 2015 (figure 2).
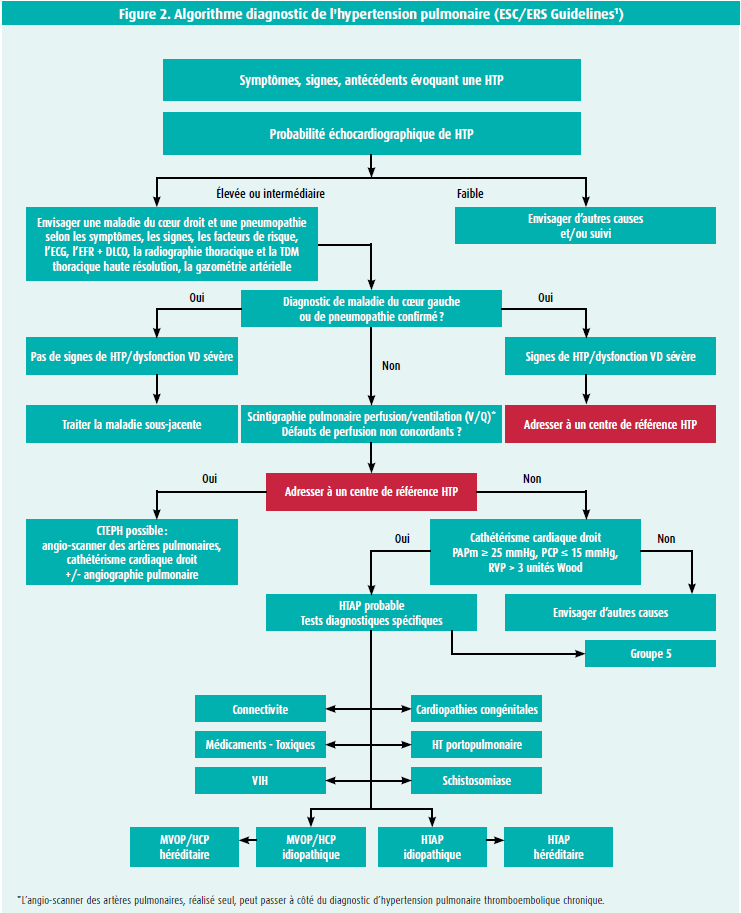

Une prise en charge diagnostic structurée permettra de raccourcir le délai entre l'apparition des premiers symptômes du patient et le diagnostic d'HTP, afin de sélectionner au plus vite les patients devant être référés vers des centres d'expertise.
Symptomatologie suspecte
En effet, l'HTP se caractérise par des symptômes très peu spécifiques, tel que de la dyspnée ou de la fatigue, et des signes cliniques de congestion droite pouvant être initialement subtiles, rendant le diagnostic difficile et souvent tardif par rapport au début d'apparition des symptômes. Ainsi, en cas de suspicion clinique d'HTP, il sera nécessaire de débuter une évaluation non invasive, incluant un électrocardiogramme, une radiographie de thorax, des épreuves fonctionnelles respiratoires et une échocardiographie transthoracique. Celle-ci permettra de déterminer la probabilité échographique d'HTP.
Echocardiographie transthoracique
L'estimation de la pression pulmonaire systolique par le jet de régurgitation tricuspide et la mise en évidence d'autres signes échographiques associés à l'HTP permettront d'établir la probabilité (faible - intermédiaire - élevée) échographique d'hypertension pulmonaire. Voici les signes échographiques complémentaires à rechercher, en faveur d'un profil hémodynamique droit :
- Morphologie ventriculaire :
- Rapport entre le diamètre basal du ventricule droit et du ventricule gauche > 1
- Aplatissement du septum interventriculaire (index d'excentricité du ventricule gauche > 1,1 en systole et/ou en diastole)
- Flux et diamètre de l'artère pulmonaire :
- Raccourcissement du temps d'accélération pulmonaire < 105 m/sec et/ ou présence d'une décélération en mésosystole
- Insuffisance pulmonaire proto-diastolique > 2,2 m/sec
- Diamètre de l'artère pulmonaire > 25 mm
- Diamètre de la veine cave inférieure et de l'oreillette droite :
- Diamètre de la veine cave inférieure > 21 mm avec un collapsus inspiratoire réduit (< 50 % à l'inspiration forcée, < 20 % à l'inspiration spontanée)
- Diamètre de l'oreillette droite (en télésystole) > 18 cm
Ces données échographiques seront ensuite associées aux données cliniques du patient : évaluer la présence ou non de facteurs de risque en faveur d'une HTAP (antécédents familiaux de HTAP, histoire personnelle de connectivite, infection par le VIH…) ou d'une CTEPH (présence de troubles de la coagulation, maladies inflammatoires chroniques, antécédents de cancers…). Ainsi, un patient symptomatique avec une probabilité échographique intermédiaire d'HTP mais présentant des facteurs de risque pour une HTAP ou une CTEPH, ou bien un patient sans facteurs de risque mais avec une haute probabilité échographique d'HTP devront être référés vers un centre d'expertise afin de compléter le bilan par un cathétérisme cardiaque droit qui permettra de confirmer ou infirmer le diagnostic (figure 3).1
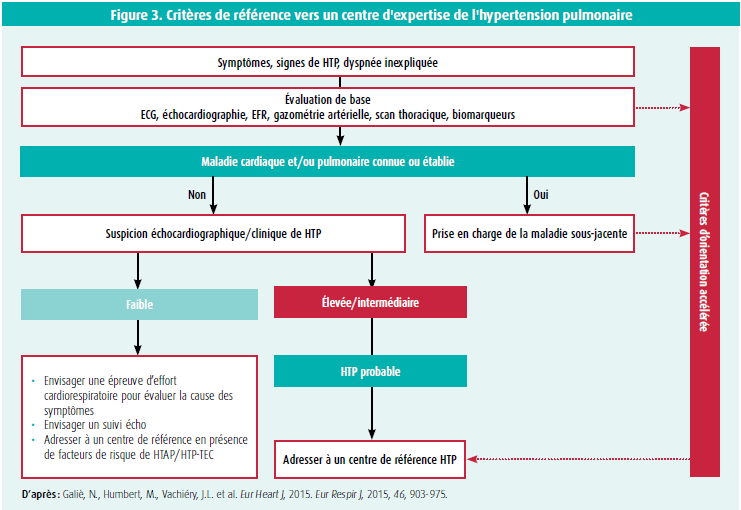
L'étape suivante sera d'exclure les formes les plus fréquentes d'HTP, que sont l'hypertension pulmonaire associée aux pathologies cardiaques gauches (groupe 2)2 et l'hypertension pulmonaire liée aux maladies respiratoires chroniques (groupe 3), constituant plus de 80 % des HTP1. La probabilité pré-test d'HTP du groupe 2 pourra être évaluée par des scores de probabilité, recherchant la présence de facteurs de risque tel que des anomalies structurelles cardiaques gauches, une fibrillation auriculaire, une dilatation auriculaire gauche, des anomalies à l'ECG (BBG, HVG), un antécédent de chirurgie cardiaque, la présence d'un syndrome métabolique, une ergospirométrie altérée avec une pente VE/VCO2 augmentée3. En cas de suspicion de CTEPH, la scintigraphie pulmonaire de ventilation et de perfusion aura tout son intérêt, car elle permettra de mettre en évidence des anomalies de perfusion caractéristiques de la maladie thrombo-embolique chronique (sensibilité de 96-97 %, spécificité de 90-95 %).
La sévérité de l'HTP pourra finalement être évaluée par le dosage des NTproBNP, la réalisation d'une IRM myocardique et par la réalisation de tests fonctionnels (test de marche de 6 minutes, ergospirométrie).
L'hypertension artérielle pulmonaire (HTAP)
L'HTP (toutes causes confondues) correspond donc à une situation hémodynamique (et non pas à une pathologie en soi), et affecte près de 450 millions de patients dans le monde. L'HTAP (groupe 1) quant à elle est une maladie rare4. Son incidence est de 2,5 à 5 adultes par millions d'habitants par an : on estime ainsi la survenue de près de 50 nouveaux cas d'HTAP par an en Belgique. Sa prévalence est de 15 à 50 adultes par millions d'habitants. En Belgique, près de 500 patients en sont atteints5. Il s'agit d'une maladie grave et incurable, avec une médiane de survie sans traitement inférieure à 3 ans. Des traitements efficaces existent, et permettent de ralentir l'évolution clinique de la maladie: les antagonistes des récepteurs de l'endothéline, les inhibiteurs de la phosphodiestérase de type 5, les stimulateurs de la guanylate cyclase et les analogues de la prostacycline ou les stimulateurs des récepteurs des prostacyclines1.
Hémodynamiquement, l'HTAP est définie par la présence d'une hypertension pulmonaire pré-capillaire (PAPm ≥ 25 mmHg et une PAPO ≤ 15 mmHg) et une PVR > 3 UW, en l'absence d'autres causes d'hypertension pulmonaire pré-capillaire, telle qu'une pathologie pulmonaire chronique, une CTEPH ou d'autres maladies rares (groupe 5)1.
Comme cité précédemment, l'HTAP est dans près de 50 % des cas d'origine idiopathique, héréditaire ou associée à une exposition à des toxiques (tel que l'aminorex, la fenfluramine, …). Le sous-groupe d'HTAP associée à d'autres conditions tel que les connectivites, l'infection par le VIH, l'hypertension portale ou les cardiopathies congénitales correspond aux 50 % des cas d'HTAP restants, avec la sclérose systémique en tête de file4.
Cas particulier d'HTAP : la sclérose systémique
La sclérose systémique est une maladie rare (prévalence de 1/10 000 adultes) et hétérogène du tissu conjonctif. Elle est caractérisée par une fibrose tissulaire diffuse, ainsi qu'une oblitération vasculaire des petits vaisseaux, d'origine auto-immune.6
La sclérose systémique peut dans certains cas se compliquer d'HTP, sur fibrose pulmonaire ou sur pathologie cardiaque gauche. Dans certains cas, on assistera à l'apparition précoce d'une HTAP7. Un screening cardiaque annuel est donc conseillé chez tout patient présentant une sclérose systémique, afin de diagnostiquer le plus précocement possible ces potentielles complications, y compris l'HTAP qui est associé à un pronostic très sévère, et d'ainsi améliorer la survie des patients à long terme8.
Centres de référence d'hypertension pulmonaire : prise en charge pluridisciplinaire et multiprofessionnelle
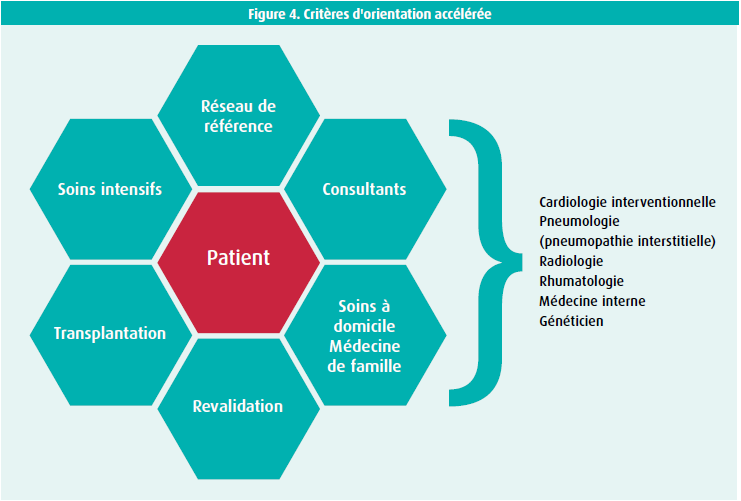
Pour garantir une prise en charge optimale de la maladie, il conseille que les patients souffrant d'HTP sévère soient pris en charge dans des centres de référence. Comme expliqué précédemment, il s'agit de maladies variées et complexes, nécessitant une prise en charge multidisciplinaire par notamment le cardiologue, le pneumologue, le rhumatologue, … afin de poser un diagnostic précis. Par la suite, il sera primordial d'assurer un suivi médical régulier, mais également un suivi multi-professionnels, impliquant de nombreux acteurs des soins de santé, tel que les infirmiers spécialisés, les kinésithérapeutes, les nutritionnistes, les assistants sociaux. Le but de ces centres de référence sera donc de regrouper les ressources, pour offrir au patient une expertise et des compétences à la pointe des connaissances actuelles.
Conclusion
En guise de conclusion, rappelons que les symptômes de l'HTP ne sont pas spécifiques et les signes cliniques associés peuvent être subtils, ce qui risque de retarder le diagnostic et donc la prise en charge de la maladie. Les étiologies de l'HTP sont variées, et l'identification de la cause exacte de l'HTP peut être laborieuse, particulièrement chez les patients âgés pouvant présenter de multiples comorbidités. L'HTP étant une condition physiopathologique fréquente, il ne faut pas la confondre avec l'HTAP qui est une entité rare et faisant l'objet de recommandations thérapeutiques précises. Selon la probabilité clinique et échocardiographique d'HTP, il sera recommandé d'adresser le patient vers un centre de référence pour l'HTP, afin qu'il puisse bénéficier d'une prise en charge optimale, combinant expérience et expertise à la pointe de l'innovation diagnostique et thérapeutique. Le but principal est de créer un réseau solide autour du patient, où chaque acteur a son importance (du médecin généraliste au spécialiste, en passant par l'ensemble des acteurs des soins paramédicaux).
Références
- Galiè, N., Humbert, M., Vachiéry J.L., Gibbs, S., Lang, I. et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J, 2015, 46 (6), 903-975. Eur Heart J, 2016, 37 (1), 67-119.
- Vachiéry, J.L., Adir, Y., Barberà, J.A., Champion, H., Coghlan, J.G. et al. Pulmonary hypertension due to heart diseases. J Am Coll Cardiol, 2013, 62, D100-108.
- Vachiéry, J.L., Tedford, R.J., Rosenkranz, S., Palazzini, M., Lang, I. et al. Pulmonary hypertension due to left heart diseases. Eur Respir J, 2019, 53 (1).
- Humbert, M., Sitbon, O., Chaouat, A., Bertocchi, M., Habib, G. et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med, 2006, 173 (9), 1023-1030.
- Belgian National Plan for Rare Diseases.
- Cutolo, M., Soldano, S., Smith, V. et al. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol, 2019, 15 (7), 753-764.
- Elhai, M., Meune, C., Boubaya, M., Avouac, J., Hachulla, E. et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis, 2017, 76 (11), 1897-1905.
- Frost, A., Badesch, D., Gibbs, J.S.R., Gopalan, D., Khanna, D. et al. Diagnosis of pulmonary hypertension. Eur Respir J, 2019, 53 (1), 1801904.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.