Symposiumverslag - EHRA-congres
Inleiding
Pulmonale hypertensie is een pathofysiologische conditie waarbij de gemiddelde druk in de longslagader in rust bij rechterhartkatheterisatie hoger is dan 25 mmHg. De klinische classificatie van pulmonale hypertensie telt vijf groepen met aandoeningen die een aantal kenmerken gemeen hebben zoals de klinische presentatie, de pathofysiologische mechanismen, de hemodynamiek en het therapeutische beleid. De frequentste vormen van pulmonale hypertensie zijn groep 2 (pulmonale hypertensie door linkerhartlijden) en groep 3 (pulmonale hypertensie door een ziekte van het ademhalingsapparaat en/of hypoxemie). Pulmonale arteriële hypertensie (groep 1) is een zeldzame, ernstige, snel progressieve en ongeneeslijke ziekte. Pulmonale arteriële hypertensie kan idiopathisch of hereditair zijn, kan te wijten zijn aan toxische stoffen en kan samenhangen met een bindweefselziekte, een hiv-infectie, portale hypertensie en een aangeboren hartziekte. Het is belangrijk die verschillende ziektebeelden van elkaar te onderscheiden. Sommige vergen immers een specifieke behandeling, meer bepaald pulmonale arteriële hypertensie en pulmonale hypertensie als gevolg van chronische trombo-embolie (groep 4) (figuur 1)1.
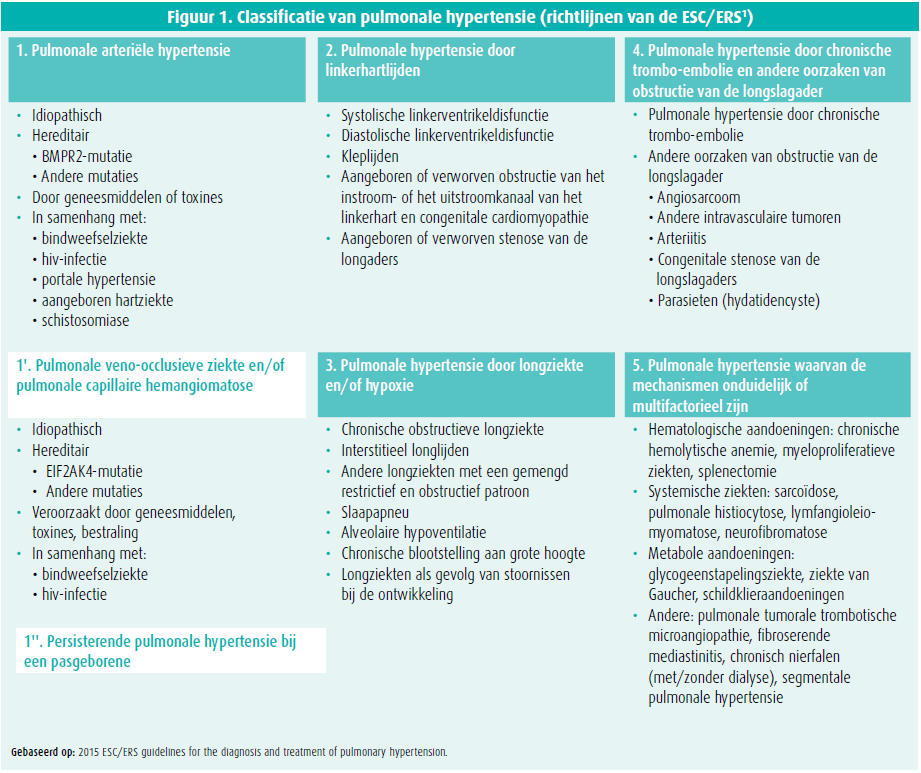
Hemodynamische definitie
De hemodynamische definitie van pulmonale hypertensie (PAPm ≥ 25 mmHg) houdt rekening met de pulmonale arteriële occlusiedruk. Als die 15 mmHg of lager is, spreken we van precapillaire pulmonale hypertensie (pulmonale hypertensie groep 1, 3, 4 en 5). Als de pulmonale wiggendruk > 15 mmHg is, spreken we van postcapillaire pulmonale hypertensie (groep 2 en 5), die verder opgesplitst wordt in geïsoleerde (IpcPH) en gecombineerde (CpcPH) hypertensie. Geïsoleerde postcapillaire pulmonale hypertensie wordt gedefinieerd als een pulmonale diastolische gradiënt (GDP = PAPd-wiggendruk) < 7 mmHg en/of een pulmonale vaatweerstand ≤ 3 UW. Gecombineerde postcapillaire pulmonale hypertensie wordt gedefinieerd als een GDP ≥ 7 mmHg en/of een pulmonale vaatweerstand > 3 UW1. Zoals al gezegd, kan een diagnose van pulmonale hypertensie enkel bevestigd worden door een rechterhartkatheterisatie, waarbij tevens de presentatie beschreven kan worden. Bij welke patiënten moet gedacht worden aan pulmonale hypertensie, waarbij ze dan verwezen moeten worden naar een expertisecentrum voor rechterhartkatheterisatie?
Diagnostisch beleid
De ESC heeft in zijn Guidelines for diagnosis and treatment of pulmonary hypertension van 2015 een diagnostisch algoritme voor pulmonale hypertensie gepubliceerd (figuur 2).
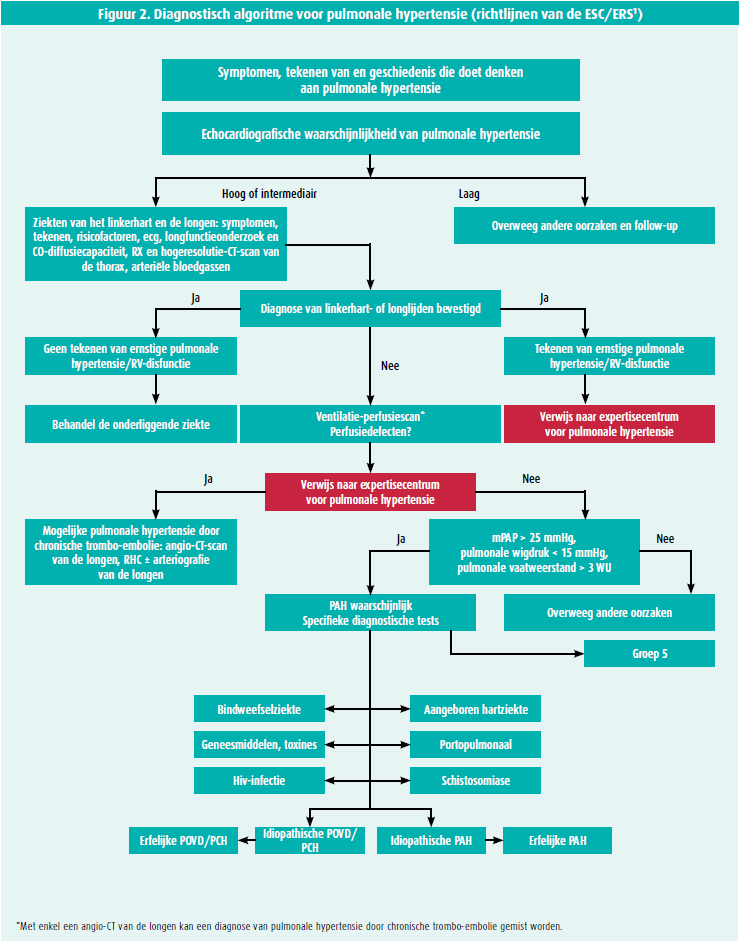

Met een gestructureerd diagnostisch beleid verkort de tijd tussen het verschijnen van de eerste symptomen en de diagnose van pulmonale hypertensie, en kunnen de patiënten sneller verwezen worden naar een expertisecentrum.
Verdachte symptomatologie
De symptomen van pulmonale hypertensie, zoals dyspneu en vermoeidheid, zijn zeer weinig specifiek en de klinische tekenen van stuwing rechts zijn aanvankelijk heel discreet, wat de differentiële diagnose bemoeilijkt en waardoor de diagnose vaak pas laat gesteld wordt. Bij klinisch vermoeden van pulmonale hypertensie is een niet-invasieve evaluatie vereist met een elektrocardiogram, een RX thorax, een longfunctieonderzoek en een transthoracale echocardiografie (voor raming van de echografische waarschijnlijkheid van pulmonale hypertensie).
Transthoracale echocardiografie
Door meting van de systolische druk in de longslagader op grond van de jet van tricuspidalis-insufficiëntie en andere echografische tekenen van pulmonale hypertensie kan men de echografische waarschijnlijkheid van pulmonale hypertensie ramen (laag - intermediair - hoog). Echografische tekenen die wijzen op een hemodynamisch profiel van het rechterhart zijn:
- Vorm van het rechterventrikel:
- Verhouding tussen de basale diameter van het rechterventrikel en die van het linkerventrikel > 1
- Afplatting van het interventriculaire septum (index van excentriciteit van het linkerventrikel > 1,1 tijdens de systole en/of de diastole)
- Flux en diameter van de longslagader:
- Verkorting van de pulmonale acceleratietijd < 105 ms en/of deceleratie in het midden van de systole
- Protodiastolische pulmonalisinsufficiëntie > 2,2 m/sec.
- Diameter van de longslagader > 25 mm
- Diameter van de vena cava inferior en het rechteratrium:
- Diameter van de vena cava inferior > 21 mm met geringere collaps tijdens inademing (< 50 % bij geforceerde inademing, < 20 % bij spontane inademing)
- Diameter van de rechtervoorkamer (op het einde van de systole) > 18 cm
Die echografische gegevens worden dan gekoppeld aan de klinische gegevens van de patiënt: al dan niet risicofactoren voor pulmonale arteriële hypertensie (familiaire voorgeschiedenis van pulmonale arteriële hypertensie, persoonlijke voorgeschiedenis van bindweefselziekte, hiv-infectie …) of voor pulmonale hypertensie door chronische trombo-embolie (stollingsstoornissen, chronische ontstekingsziekte, voorgeschiedenis van kanker …). Een patiënt met symptomen, een intermediaire echografische waarschijnlijkheid van pulmonale hypertensie en risicofactoren voor pulmonale hypertensie of pulmonale hypertensie door chronische trombo-embolie of een patiënt zonder risicofactoren, maar met een hoge echocardiografische waarschijnlijkheid van pulmonale hypertensie, moet naar een expertisecentrum verwezen worden voor rechterhartkatheterisatie om de diagnose te bevestigen of te ontkrachten (figuur 3).1
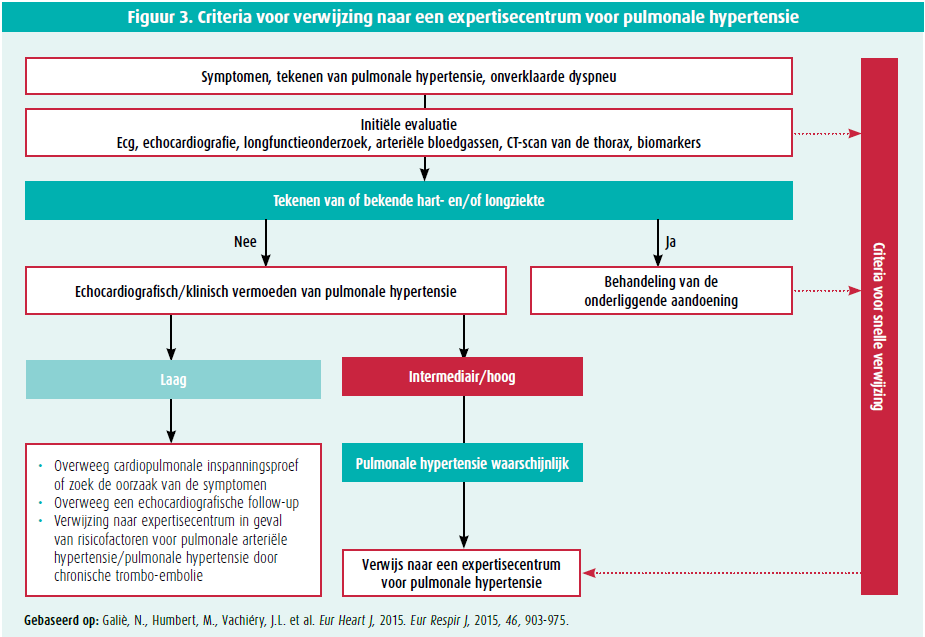
Daarna moeten de frequentste oorzaken van pulmonale hypertensie uitgesloten worden, namelijk pulmonale hypertensie in samenhang met linkerhartlijden (groep 2)2 en pulmonale hypertensie als gevolg van een chronische ademhalingsziekte (groep 3) (die zijn samen goed voor meer dan 80 % van de gevallen van pulmonale hypertensie).1 De pretestwaarschijnlijkheid van pulmonale hypertensie van groep 2 kan geëvalueerd worden met waarschijnlijkheidsscores die gebaseerd zijn op risicofactoren, zoals structurele afwijkingen van het linkerhart, atriumfibrillatie, verwijding van het linkeratrium (ecg-afwijkingen (VLBTB, LVH), voorgeschiedenis van hartchirurgie, metabool syndroom, abnormale resultaten bij ergospirometrie met een steilere VE/VCO2-helling.3 Bij vermoeden van pulmonale hypertensie door chronische trombo-embolie wordt een ventilatie- perfusiescintigrafie van de longen uitgevoerd om de kenmerkende perfusiedefecten bij chronische trombo-embolie op te sporen (sensitiviteit van 96-97 % en specificiteit van 90-95 %).
De ernst van de pulmonale hypertensie wordt beoordeeld door bepaling van de NT-proBNP-spiegel, een MRI van het myocard en functionele tests (6 minutenwandeltest, ergospirometrie).
Pulmonale arteriële hypertensie (PAH)
Pulmonale hypertensie (ongeacht de oorzaak) is een hemodynamische conditie (en dus niet een ziekte als dusdanig). Wereldwijd vertonen bijna 450 miljoen mensen pulmonale hypertensie. Pulmonale arteriële hypertensie (PAH) (groep 1) is een zeldzame ziekte.4 De incidentie wordt geraamd op 2,5-5 volwassenen per miljoen inwoners per jaar. In België wordt de incidentie van pulmonale arteriële hypertensie geraamd op bijna 50 gevallen per jaar. De prevalentie bedraagt 15-50 volwassenen per miljoen inwoners. In België vertonen bijna 500 patiënten pulmonale arteriële hypertensie.5 Het is een ernstige, ongeneeslijke ziekte. De mediane overleving zonder behandeling is korter dan 3 jaar. Er bestaan doeltreffende behandelingen, waarmee de klinische evolutie van de ziekte afgeremd kan worden: endothelinereceptorantagonisten, type 5-fosfodi-esteraseremmers, guanylaatcyclase-stimulerende middelen, prostacyclineanalogen en stoffen die prostacyclinereceptor stimuleren.1
Hemodynamisch wordt pulmonale arteriële hypertensie gedefinieerd als een precapillaire pulmonale hypertensie (PAPm ≥ 25 mmHg en pulmonale arteriële occlusiedruk ≤ 15 mmHg) en een pulmonale vaatweerstand > 3 UW zonder andere oorzaken van precapillaire pulmonale hypertensie, zoals chronisch longlijden, pulmonale hypertensie door chronische trombo-embolie en andere zeldzame ziekten (groep 5)1
Bijna 50 % van de gevallen van pulmonale arteriële hypertensie is idiopathisch, hereditair of te wijten aan toxische stoffen (zoals aminorex, fenfluramine …). De overige 50 % hangt samen met een andere ziekte, zoals een bindweefselziekte, hiv-infectie, portale hypertensie of een aangeboren ziekte. De frequentste oorzaak is systemische sclerose.4
Bijzonder geval van pulmonale arteriële hypertensie: systemische sclerose
Systemische sclerose (sclerodermie) is een zeldzame (prevalentie 1/10 000 volwassenen) en heterogene auto-immuunziekte van het bindweefsel, die gekenmerkt wordt door diffuse weefselfibrose en occlusie van de kleine bloedvaatjes.6
Een mogelijke complicatie van sclerodermie is pulmonale hypertensie als gevolg van longfibrose of linkerhartlijden. Soms treedt snel pulmonale arteriële hypertensie op.7 Een jaarlijks hartonderzoek is dus wenselijk bij patiënten met sclerodermie, om complicaties zoals pulmonale arteriële hypertensie (de prognose is dan zeer slecht) op te sporen en de langetermijnoverleving van de patiënten te verbeteren.8
Referentiecentra voor pulmonale hypertensie: multidisciplinair en multiprofessioneel beleid
Het is raadzaam patiënten met een ernstige pulmonale hypertensie te behandelen in een referentiecentrum om een optimale aanpak te verzekeren. Het gaat immers om uiteenlopende en complexe aandoeningen die een multidisciplinair beleid vergen (cardioloog, pneumoloog, reumatoloog …) om een precieze diagnose te stellen. Daarna moet de patiënt regelmatig gevolgd worden door artsen en andere gezondheidswerkers zoals gespecialiseerde verpleegkundigen, kinesitherapeuten, voedingsdeskundigen en maatschappelijk werkers. Die referentiecentra bundelen de vereiste expertise en competenties.
Conclusie
De symptomen van pulmonale hypertensie zijn niet specifiek en de klinische tekenen kunnen subtiel zijn. Daardoor dreigt de diagnose pas laat gesteld te worden, zodat de patiënt niet tijdig een behandeling krijgt. Er zijn tal van oorzaken van pulmonale hypertensie. Het is niet altijd eenvoudig de juiste oorzaak van pulmonale hypertensie te achterhalen, vooral bij oudere patiënten, die vaak meerdere comorbiditeiten vertonen. Pulmonale hypertensie is een frequente pathofysiologische conditie, die echter niet verward mag worden met pulmonale arteriële hypertensie. PAH is een zeldzame entiteit, waarvoor specifieke therapeutische richtlijnen bestaan. Naargelang de klinische en echocardiografische waarschijnlijkheid van pulmonale hypertensie wordt aanbevolen de patiënt te verwijzen naar een referentiecentrum voor pulmonale hypertensie, met het oog op een optimale aanpak dankzij ervaring, expertise en kennis van de diagnostische en therapeutische nieuwigheden. Het hoofddoel is een stevig netwerk te vormen rond de patiënt, waarbij alle betrokkenen belangrijk zijn: huisarts, specialist en paramedici.
Referenties
- Galiè, N., Humbert, M., Vachiéry J.L., Gibbs, S., Lang, I. et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J, 2015, 46 (6), 903-975. Eur Heart J, 2016, 37 (1), 67-119.
- Vachiéry, J.L., Adir, Y., Barberà, J.A., Champion, H., Coghlan, J.G. et al. Pulmonary hypertension due to heart diseases. J Am Coll Cardiol, 2013, 62, D100-108.
- Vachiéry, J.L., Tedford, R.J., Rosenkranz, S., Palazzini, M., Lang, I. et al. Pulmonary hypertension due to left heart diseases. Eur Respir J, 2019, 53 (1).
- Humbert, M., Sitbon, O., Chaouat, A., Bertocchi, M., Habib, G. et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med, 2006, 173 (9), 1023-1030.
- Belgian National Plan for Rare Diseases.
- Cutolo, M., Soldano, S., Smith, V. et al. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol, 2019, 15 (7), 753-764.
- Elhai, M., Meune, C., Boubaya, M., Avouac, J., Hachulla, E. et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis, 2017, 76 (11), 1897-1905.
- Frost, A., Badesch, D., Gibbs, J.S.R., Gopalan, D., Khanna, D. et al. Diagnosis of pulmonary hypertension. Eur Respir J, 2019, 53 (1), 1801904.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.