L'insuffisance cardiaque à fraction d'éjection réduite est une pathologie rare chez les sujets jeunes. Bien que la première hypothèse soit une cardiomyopathie non ischémique, génétiquement déterminée, nous rapportons un cas de malformation coronaire à l'origine d'une insuffisance cardiaque sévère chez un sujet jeune.
La malformation ALCAPA implique une origine aberrante de l'artère coronaire gauche au départ de l'artère pulmonaire au lieu de l'aorte. Dans ce cas, dès la naissance, le ventricule gauche est alimenté en sang désoxygéné, ce qui peut s'accompagner d'un syndrome de Bland-White-Garland (combinaison d'une insuffisance cardiaque et d'un retard de croissance). Le diagnostic est généralement posé au cours de la première année de vie, suite à l'apparition d'une insuffisance cardiaque, et dans certains cas après une mort subite.
Le traitement consiste à réimplanter chirurgicalement le tronc commun sur l'aorte, ou à greffer la LIMA sur l'IVA proximale et à ligaturer le tronc commun. On sait encore peu de choses sur le résultat fonctionnel à long terme.
Introduction
Les malformations coronaires, rares en pratique quotidienne, sont principalement représentées par un trajet aberrant des coronaires, une fistulisation ou des ponts myocardiques. L'origine anormale des coronaires fait partie des malformations congénitales plus rares, l'origine anormale de l'artère coronaire gauche (LCA) sur l'artère pulmonaire (AP) étant considérée comme l'une des plus dangereuses.1 Nous discutons du cas d'un jeune sportif chez qui ce diagnostic a été posé à l'âge de 17 ans.
Cas clinique
Le patient, un jeune homme de 17 ans sans antécédents médicaux, s'est présenté pour la première fois en consultation de Cardiologie en raison d'une dyspnée à l'effort. Trois mois auparavant, il jouait encore au football en compétition, mais il avait remarqué qu'il était plus vite essoufflé que ses partenaires de jeu. Ces derniers temps, il avait remarqué qu'il était plus vite dyspnéique et tachycarde lorsqu'il faisait un effort. Les symptômes avaient nettement augmenté au cours des 3 derniers mois. On ne notait pas d'antécédents familiaux.
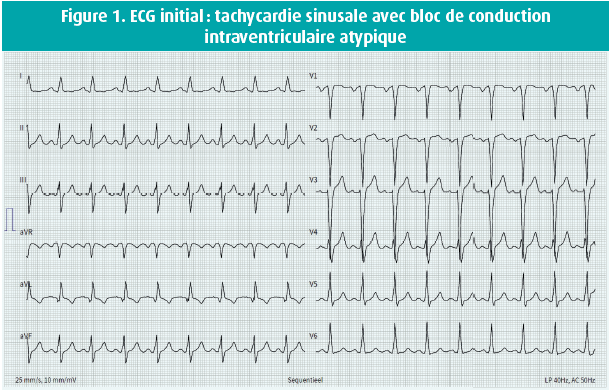
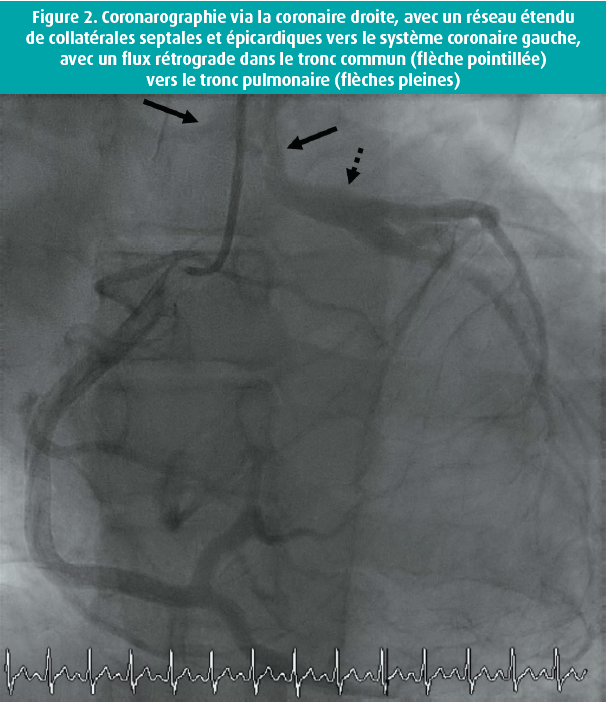
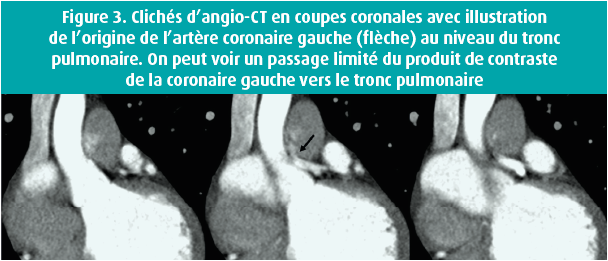
L'examen clinique était normal, hormis un léger souffle systolique. L'ECG montrait une tachycardie sinusale avec un bloc de conduction intraventriculaire atypique (figure 1). À l'échocardiographie, nous avons vu un ventricule gauche dilaté dont la fonction était fortement réduite (fraction d'éjection de 28 %), une insuffisance mitrale sévère avec dilatation biauriculaire et une hypertension pulmonaire (pression pulmonaire systolique calculée de 63 mmHg). Un cathétérisme gauche-droit complémentaire a montré une origine aberrante de la LCA au niveau de l'AP. La perfusion myocardique était assurée via l'artère coronaire droite, bien développée, avec un réseau étendu de collatérales septales et épicardiques vers le système coronaire gauche, avec un flux rétrograde dans le tronc commun vers l'AP (figure 2). Une réévaluation du CT scan coronaire a confirmé ce diagnostic (figure 3). Une IRM n'a pas montré de captation pathologique focale ni de signes de fibrose. On a lancé un panel génétique complémentaire, qui n'a pas révélé d'anomalies significatives.
On a instauré un traitement de l'insuffisance cardiaque au moyen de bêtabloquants, IEC, spironolactone et diurétiques de l'anse, et le patient a été adressé à un centre spécialisé en vue d'une chirurgie. Toutefois, peu avant l'intervention planifiée, le patient a noté une dégradation de son état général, avec dyspnée et fatigue croissantes, de sorte qu'il a finalement été opéré plus tôt que prévu.
On a pratiqué une réimplantation du tronc commun sur l'aorte ainsi qu'une annuloplastie de la valve mitrale. Les suites postopératoires ont été caractérisées par une nette dégradation de la fonction ventriculaire gauche, l'apparition d'une fibrillation auriculaire et la nécessité d'un soutien inotrope au moyen de lévosimendan. Lors d'une consultation de contrôle 2 mois après l'intervention, le ventricule gauche restait modérément dilaté, avec une fonction inchangée comparativement à la valeur préopératoire, et on notait une trace d'insuffisance mitrale résiduelle et une normalisation des pressions droites. Cliniquement, le patient notait une nette amélioration de sa capacité à l'effort.
Discussion
La malformation ALCAPA (anomalous left coronary artery originating from the pulmonary artery) est une malformation coronaire rare lors de laquelle la LCA est incorrectement implantée sur l'AP. Cette anomalie a été décrite pour la première fois en 1911 lors d'une autopsie.² L'incidence est estimée à 1 sur 300 000 naissances vivantes,3 sur la base des données du Toronto Heart Registry de 1959.4 Une étude plus récente réalisée en 1990 a relevé une incidence de 0,008 % parmi tous les patients subissant une coronarographie.5
Cliniquement, l'ALCAPA a une présentation hétérogène. Les patients peuvent notamment présenter un souffle cardiaque continu, une angine de poitrine, un infarctus myocardique, une insuffisance cardiaque congestive, des troubles du rythme ou une mort subite. Le plus typique est l'apparition de signes d'insuffisance cardiaque après les premiers mois de vie, ce qui a été décrit en 1933 sous le nom de syndrome de Bland-White-Garland.6 Dans la première moitié du XXe siècle, l'insuffisance cardiaque et la mort subite ont été décrites comme la principale présentation clinique, 90 % des patients décédant avant l'âge d'un an. Au cours des dernières décennies, le diagnostic a également été posé à plusieurs reprises chez des patients adultes, vraisemblablement en raison de la disponibilité croissante de différents outils diagnostiques. Une revue de la littérature de Yau et al. a compilé 151 rapports de cas de malformation ALCAPA chez des adultes, publiés entre 1908 et 2008, l'âge moyen au moment du diagnostic étant de 41 ans.7 Dans ces cas, 66 % des patients présentaient des plaintes de type dyspnée, palpitations ou fatigue, 17 % une syncope, des arythmies ventriculaires ou une mort subite. 14 % étaient asymptomatiques. Les 12 % restants constituaient des observations post-mortem à l'autopsie.
Les facteurs qui déterminent la survie jusqu'à un âge plus avancé ne sont pas clairs. On peut supposer que le degré de développement de la circulation collatérale de l'artère coronaire droite vers le système gauche joue un rôle important à cet égard.7 Initialement, on suppose que le sang désoxygéné parvient au ventricule gauche via un flux antérograde à travers le tronc commun, suite à quoi la circulation collatérale se développe. Une fois qu'il y a suffisamment de collatérales, il y a une transition vers un flux rétrograde à travers le tronc commun et un shunt vers l'AP. Cependant, ceci est progressivement associé à un syndrome de 'vol' coronaire en raison de la baisse des pressions dans la circulation pulmonaire, ce qui peut entraîner une baisse des pressions de perfusion dans toute la circulation coronaire. Le développement concomitant d'une hypertension pulmonaire consécutive à une insuffisance cardiaque (comme c'est le cas ici) peut constituer un mécanisme compensatoire non intentionnel, susceptible de maintenir la pression de perfusion coronaire. Il s'agit toutefois d'une hypothèse - bien qu'elle puisse expliquer pourquoi l'état clinique de notre patient s'est aggravé après l'instauration des diurétiques de l'anse, entre autres.
La chirurgie, visant à rétablir la double perfusion coronaire, est la pierre angulaire du traitement. Dans des séries de cas plus anciennes, on a tenté d'augmenter la pression de perfusion coronaire en ligaturant sélectivement le tronc commun.8 Cependant, cela n'a pas réduit l'incidence des morts subites, contrairement à la ligature combinée du tronc commun et à la pose d'un greffon veineux sur l'IVA.9 Une autre forme de chirurgie est la réparation dite de Takeuchi, qui consiste à construire une fenêtre aorto-pulmonaire et à creuser un tunnel entre cette fenêtre et l'ostium de la LCA, à l'intérieur du tronc pulmonaire. En raison de la complexité de l'intervention et du risque de développer une sténose de l'artère pulmonaire, cette pratique a été abandonnée.10,11 Enfin, le rétablissement de la double perfusion coronaire est recommandé, de préférence via la réimplantation de la LCA sur l'aorte et, si ce n'est pas possible, via la ligature de la LCA et la réalisation d'un pontage.
La récupération fonctionnelle après un traitement chirurgical semble possible à long terme. Ceci est principalement basé sur des études portant sur des patients de moins d'un an.10 De petites séries de cas montrent des résultats positifs provisoires sur le plan de la récupération de la fraction d'éjection du ventricule gauche après une correction chirurgicale à l'âge adulte.11,12 On sait peu de choses sur le pronostic à long terme de la malformation ALCAPA, principalement en raison de la faible prévalence de la malformation, mais aussi de l'hétérogénéité des populations de patients étudiées.
Conclusion
L'ALCAPA est une malformation congénitale grave, rare, de présentation hétérogène. Typiquement, elle est constatée au cours des premiers mois de vie, ou post-mortem après une mort subite du nourrisson. Dans de rares cas, le diagnostic est posé à l'âge adulte, soit fortuitement, soit après l'apparition précoce d'une insuffisance cardiaque. Une réparation chirurgicale visant à rétablir la double perfusion coronaire est toujours indiquée. Le pronostic à long terme après une réparation chirurgicale semble favorable.
Références
- Pérez-Pomares, J.M., de la Pompa, J.L., Franco, D., Sedmera, D., Sheppard, M., Sperling, S. et al. Congenital coronary artery anomalies: a bridge from embryology to anatomy and pathophysiology—a position statement of the development, anatomy, and pathology ESC Working Group. Cardiovasc Res, 2016, 109 (2), 204-2016.
- Abrikosoff, A. Arch. path. Anat, 1911, 203, 413.
- Dodge-Khatami, A., Mavroudis, C., Backer, C.L. Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery: Collective Review of Surgical Therapy. Ann Thorac Surg, 2002, 74 (3), 946-955.
- Keith, J. The anomalous origin of the left coronary artery from the pulmonary artery. Br Heart J, 1959, 21, 149-161.
- Yamanaka, O., Hobbs, R. Coronary Artery Anomalies in 126,595 Patients Undergoing Coronary Arteriography. Cathet Cardiovasc Diagn, 1990, 21 (1), 28-40.
- Bland, E.F., White, P.D., Garland, J. Congenital anomalies of the coronary arteries: Report of an unusual case associated with cardiac hypertrophy. Am Heart J, 1933, 8, 787-801.
- Yau, J.M., Singh, R., Halpern, E.J., Fischman, D. Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery in Adults: A Comprehensive Review of 151 Adult Cases and A New Diagnosis in a 53-Year-Old Woman. Clin Cardiol, 2011, 34 (4), 204-210.
- Sabiston, D.C. Jr, Neill, C.A., Taussig, H.B. The Direction of Blood Flow in Anomalous Left Coronary Artery Arising from the Pulmonary Artery. Circulation, 1960, 22, 591-597.
- Wilson, C.L., Dlabal, P.W., McGuire, S.A. Surgical treatment of anomalous left coronary artery from pulinonary artery: Follow-up in teenagers and adults. Am Heart J, 1979, 98 (4), 440-446.
- Cochrane, A.D., Coleman, D.M., Davis, A.M., Brizard, C.P., Wolfe, R., Karl, T.R. Excellent long-term functional outcome after an operation for anomalous left coronary artery from the pulmonary artery. J Thorac Cardiovasc Surg, 1999, 117 (2), 332-342.
- Lange, R., Vogt, M., Hörer, J., Cleuziou, J., Menzel, A., Holper, K. et al. Long-Term Results of Repair of Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery. Ann Thorac Surg, 2007, 83 (4), 1463-1471.
- Sasikumar, D., Dharan, B.S., Arunakumar, P., Gopalakrishnan, A., Sivasankaran, S., Krishnamoorthy, K.M. The outcome of mitral regurgitation after the repair of anomalous left coronary artery from the pulmonary artery in infants and older children. Interact Cardiovasc Thorac Surg, 2018, 27 (2), 238-242.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.