Hartfalen met gereduceerde ejectiefractie is een zeldzame pathologie op jonge leeftijd. Hoewel dit in eerste plaats verdacht is voor nietischemische, genetisch bepaalde cardiomyopathie, rapporteren wij een geval van een coronaire malformatie als oorzaak van ernstig hartfalen op jonge leeftijd.
De ALCAPA-malformatie omvat een aberrante origine van de linkerkransslagader vanuit de arteria pulmonalis, in plaats van de aorta. Hierbij wordt vanaf de geboorte zuurstofarm bloed aangeboden aan het linkerventrikel, wat gepaard kan gaan met een Bland-White-Garlandsyndroom (een combinatie van hartfalen en failure to thrive). Meestal wordt de diagnose gedurende het eerste levensjaar gesteld bij het ontwikkelen van hartfalen, in sommige gevallen na plotse dood.
De behandeling bestaat uit een heelkundige herimplantatie van de hoofdstam op de aorta, of door het aanleggen van een LIMA-greffe op de proximale LAD en afbinden van de hoofdstam. Er is nog weinig geweten over de functionele uitkomst op lange termijn.
Inleiding
Coronaire malformaties zijn een zeldzaam gegeven in de dagelijkse praktijk, voornamelijk vertegenwoordigd door aberrant verloop van de kransslagaders, fistulisatie of myocardbruggen. Abnormale origine van de coronairen valt onder de meer zeldzame congenitale afwijkingen, waarbij abnormale origine van de linker coronaire arterie (LCA) op de arteria pulmonalis (AP) als een van de gevaarlijkste wordt beschouwd.1 We bespreken een casus van een jonge sporter, waarbij deze diagnose op 17-jarige leeftijd gesteld werd.
Klinische casus
De patiënt, een 17-jarige jongeman zonder medische voorgeschiedenis, presenteerde zich voor het eerst via de raadpleging Cardiologie naar aanleiding van inspanningsdyspneu. Tot drie maanden voordien speelde hij nog voetbal in competitief verband, waarbij werd opgemerkt dat hij sneller dan zijn leeftijdsgenoten buiten adem was. Bij inspanningen merkte hij de laatste tijd sneller kortademig en tachycard te worden. De laatste drie maanden zijn de klachten duidelijk toegenomen. Er is geen familiale voorgeschiedenis.
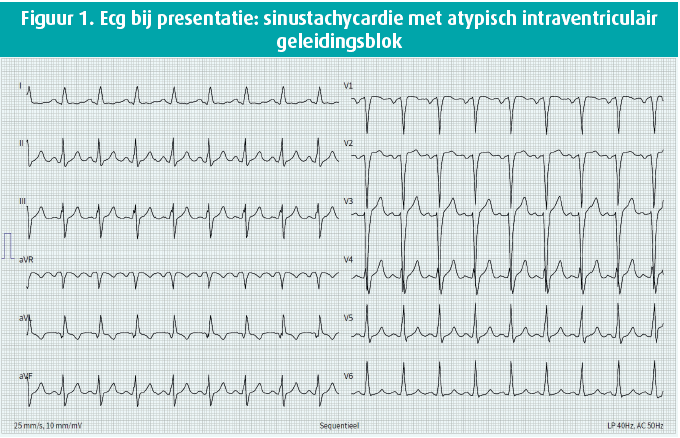
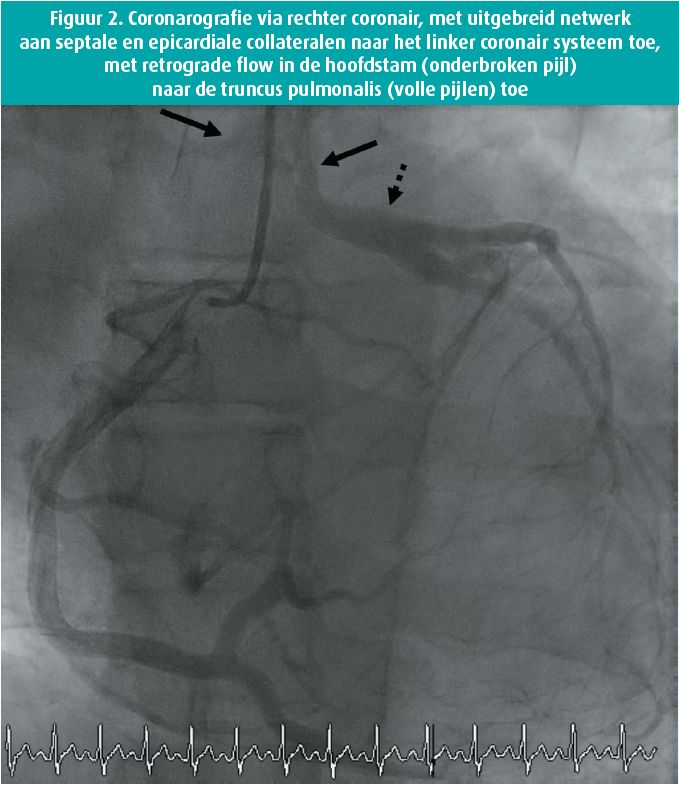
Bij fysiek onderzoek viel een mild systolische souffle op, verder was dit normaal. Het ecg toonde een sinustachycardie met atypisch intraventriculair geleidingsblok (figuur 1). Echocardiografisch zagen we een gedilateerd linkerventrikel met sterk gedaalde functie (ejectiefractie van 28 %), een ernstige mitralisklepinsufficientie met biatriale dilatatie en pulmonale hypertensie (berekende systolische pulmonaaldruk van 63 mmHg). Een aanvullende links-rechtskatheterisatie toonde een aberrante origine van de LCA ter hoogte van de AP. Myocardperfusie werd voorzien via de goed ontwikkelde rechter coronaire arterie, met een uitgebreid netwerk aan septale en epicardiale collateralen naar het linker coronair systeem toe, met retrograde flow in de hoofdstam naar de AP toe (figuur 2). Herevaluatie van de coronaire CT-scan bevestigde deze diagnose (figuur 3). Een MRI toonde geen focale pathologische captatie of tekens van fibrose. Een aanvullend genetisch panel werd gelanceerd. Dit toonde geen significante afwijkingen.

Er werd gestart met hartfalentherapie onder de vorm van bètablokkade, ACE-inhibitie, spironolacton en lisdiuretica, en de patiënt werd verwezen naar een gespecialiseerd centrum voor heelkunde. Kort voor de geplande ingreep bemerkte de patiënt echter achteruitgang van de algemene toestand, met toenemende dyspneu en vermoeidheid, waarvoor hij finaal vroeger dan voorzien voor heelkunde werd opgenomen.
Er werd een herimplantatie van de hoofdstam op de aorta, alsook annuloplastiek van de mitralisklep uitgevoerd. Het postoperatieve verloop was verwikkeld met sterke achteruitgang van de linkerventrikelfunctie, ontwikkeling van voorkamerfibrillatie en nood aan inotrope ondersteuning met levosimendan. Bij een controleconsultatie twee maanden postoperatief bleef het linkerventrikel matig gedilateerd met onveranderde functie ten opzichte van voorafgaand aan de heelkunde, met slechts een spoor residuele mitralisklepinsufficiëntie en normalisatie van de rechtszijdige drukken. Klinisch merkte de patiënt een duidelijke verbetering van de inspanningscapaciteit op.
Bespreking
ALCAPA (anomalous left coronary artery originating from the pulmonary artery) is een zeldzame coronaire malformatie waarbij de LCA verkeerdelijk geïmplanteerd is op de AP. Deze afwijking werd voor het eerst in 1911 beschreven op autopsie.2 De incidentie wordt geschat op 1 op de 300 000 levend geboren kinderen,3 gebaseerd op gegevens van de Toronto Heart Registry van 1959.4 Een meer recente studie in 1990 zag een incidentie van 0,008 % onder alle patiënten die een coronarografie ondergaan.5
Op klinisch vlak kent ALCAPA een heterogene presentatie. Patiënten kunnen zich onder andere presenteren met een continu hartgeruis, angor, myocardinfarct, congestief hartfalen, ritmestoornissen, of plotse dood. Het meest typische is het ontstaan van tekens van hartfalen na de eerste levensmaanden, wat in 1933 werd omschreven als het Bland-White-Garlandsyndroom.6 In de eerste helft van de twintigste eeuw werd hartfalen en plotse dood als de voornaamste klinische presentatie beschreven, waarbij 90 % van de patiënten overlijden voor de leeftijd van 1 jaar. In de laatste decennia werd de diagnose ook herhaaldelijk bij volwassen patiënten gesteld, vermoedelijk als gevolg van toenemende beschikbaarheid van verschillende diagnostische middelen. Een literatuurstudie van Yau et al. verzamelde 151 case reports van de ALCAPA-malformatie bij volwassenen, gepubliceerd tussen 1908 en 2008, waarbij de gemiddelde leeftijd bij diagnose 41 jaar bedroeg.7 Hierbij presenteerde 66 % van de patiënten zich met klachten van dyspneu, palpitaties, vermoeidheid of dyspneu, 17 % met syncope, ventriculaire aritmie of plotse dood. 14 % was asymptomatisch. De overige 12 % betreft post-mortem bevindingen op autopsie.
Het is onduidelijk welke factoren de overleving tot latere leeftijd bepalen. Vermoedelijk speelt de mate van ontwikkeling van collaterale circulatie van de rechter coronaire arterie naar het linkersysteem een belangrijke rol hierbij.7 Initieel wordt vermoedelijk via antegrade flow door de hoofdstam zuurstofarm bloed aan het linkerventrikel voorzien, waarbij zich collaterale circulatie ontwikkelt. Eens er voldoende collateralen gevormd zijn, volgt de overgang naar retrograde flow door de hoofdstam en shunting naar de AP. Hierbij ontstaat echter ook progressief een coronair 'steal' syndroom door de lagere drukken in de pulmonale circulatie, waarbij lagere perfusiedruk over de gehele coronaire circulatie kan ontstaan. Het concomitant ontstaan van pulmonale hypertensie als gevolg van hartfalen (zoals bij deze casus het geval is) kan er een onopzettelijk compensatiemechanisme vormen, wat de coronaire perfusiedruk in stand kan houden. Dit is echter speculatief - hoewel het een verklaring vormt voor waarom de klinische toestand van onze patiënt na het opstarten van o.a. lisdiuretica, achteruitging.
Heelkunde, gericht op herstel van duale coronaire perfusie, is de hoeksteen van de behandeling. In oudere case series werd door selectief afbinden van de hoofdstam geprobeerd de coronaire perfusiedruk te verhogen.8 Desondanks bleek dit de incidentie van plotse dood niet te verminderen, waar dit bij gecombineerd afbinden van de hoofdstam en aanleggen van een veneuze greffe naar de LAD wél het geval was.9 Een andere vorm van heelkunde is het zogenaamde Takeuchi-herstel, waarbij een aortopulmonaal venster werd aangelegd met tunneling van dit venster naar het ostium van de LCA, binnenin de truncus pulmonalis. Vanwege de complexiteit van de ingreep, en het risico op ontstaan van arteria pulmonalisstenose, werd deze procedure verlaten.10,11 Ten slotte wordt herstel van duale coronaire perfusie aanbevolen, bij voorkeur door herimplantatie van de LCA op de aorta, en als dit niet mogelijk is, door afbinden van de LCA en aanleggen van een bypassgreffe.
Functioneel herstel na chirurgische behandeling lijkt mogelijk te zijn op lange termijn. Dit is voornamelijk gebaseerd op studies bij patiënten jonger dan één jaar.10 Kleine case series tonen voorzichtig positieve resultaten over herstel van linkerventrikelejectiefractie na heelkundige correctie op volwassen leeftijd.11,12 Er is weinig geweten over de langetermijnprognose van de ALCAPA-malformatie, voornamelijk door de lage prevalentie van de malformatie, maar ook door de heterogeniteit van de patiëntenpopulaties die onderzocht zijn.
Conclusie
ALCAPA is een zeldzame, ernstige congenitale malformatie met heterogene presentatie. Typisch wordt dit vastgesteld in de eerste levensmaanden, of post-mortem na wiegedood. In zeldzame gevallen wordt de diagnose op volwassen leeftijd gesteld, hetzij toevallig, hetzij na vroegtijdige aanvang van hartfalen. Chirurgisch herstel met als doel het herstellen van duale coronaire perfusie is steeds aangewezen. De prognose op lange termijn na chirurgisch herstel lijkt gunstig.
Referenties
- Pérez-Pomares, J.M., de la Pompa, J.L., Franco, D., Sedmera, D., Sheppard, M., Sperling, S. et al. Congenital coronary artery anomalies: a bridge from embryology to anatomy and pathophysiology—a position statement of the development, anatomy, and pathology ESC Working Group. Cardiovasc Res, 2016, 109 (2), 204-2016.
- Abrikosoff, A. Arch. path. Anat, 1911, 203, 413.
- Dodge-Khatami, A., Mavroudis, C., Backer, C.L. Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery: Collective Review of Surgical Therapy. Ann Thorac Surg, 2002, 74 (3), 946-955.
- Keith, J. The anomalous origin of the left coronary artery from the pulmonary artery. Br Heart J, 1959, 21, 149-161.
- Yamanaka, O., Hobbs, R. Coronary Artery Anomalies in 126,595 Patients Undergoing Coronary Arteriography. Cathet Cardiovasc Diagn, 1990, 21 (1), 28-40.
- Bland, E.F., White, P.D., Garland, J. Congenital anomalies of the coronary arteries: Report of an unusual case associated with cardiac hypertrophy. Am Heart J, 1933, 8, 787-801.
- Yau, J.M., Singh, R., Halpern, E.J., Fischman, D. Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery in Adults: A Comprehensive Review of 151 Adult Cases and A New Diagnosis in a 53-Year-Old Woman. Clin Cardiol, 2011, 34 (4), 204-210.
- Sabiston, D.C. Jr, Neill, C.A., Taussig, H.B. The Direction of Blood Flow in Anomalous Left Coronary Artery Arising from the Pulmonary Artery. Circulation, 1960, 22, 591-597.
- Wilson, C.L., Dlabal, P.W., McGuire, S.A. Surgical treatment of anomalous left coronary artery from pulinonary artery: Follow-up in teenagers and adults. Am Heart J, 1979, 98 (4), 440-446.
- Cochrane, A.D., Coleman, D.M., Davis, A.M., Brizard, C.P., Wolfe, R., Karl, T.R. Excellent long-term functional outcome after an operation for anomalous left coronary artery from the pulmonary artery. J Thorac Cardiovasc Surg, 1999, 117 (2), 332-342.
- Lange, R., Vogt, M., Hörer, J., Cleuziou, J., Menzel, A., Holper, K. et al. Long-Term Results of Repair of Anomalous Origin of the Left Coronary Artery From the Pulmonary Artery. Ann Thorac Surg, 2007, 83 (4), 1463-1471.
- Sasikumar, D., Dharan, B.S., Arunakumar, P., Gopalakrishnan, A., Sivasankaran, S., Krishnamoorthy, K.M. The outcome of mitral regurgitation after the repair of anomalous left coronary artery from the pulmonary artery in infants and older children. Interact Cardiovasc Thorac Surg, 2018, 27 (2), 238-242.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.