Compte rendu du congrès de la BSC - session 24
Lors du congrès de la Société belge de cardiologie (BSC) de 2023, le Belgian Council on Cardiogenomics a pu organiser une session dédiée au rôle de la génétique en cardiologie. Durant cette session, les différents présentateurs ont rappelé les implications et l'importance des tests génétiques en cardiologie, notamment pour préciser le diagnostic clinique, mais aussi, dans certains cas, pour affiner le pronostic et adapter le traitement. Les tests génétiques permettent en outre d'identifier précocement les membres de la famille présentant un risque de développer une pathologie, justifiant la mise en place d'un suivi régulier. Les différents résultats qui peuvent être obtenus lors de la réalisation d'un test génétique ont également été passés en revue, de même que la démarche pour interpréter et classer un variant génétique. Dans cette approche, le conseil génétique occupe un rôle central, offrant un cadre pour discuter des limites des tests réalisés, expliquer les implications du résultat (pour le patient et les membres de sa famille) et en évaluer l'impact psychologique.
Tests génétiques dans le cadre des maladies cardiaques : état de l'art d'un domaine en constante évolution
James Ware - Imperial College, Londres, RU
La cardiogénétique est une discipline en plein essor. Un document de consensus d'experts européens a d'ailleurs été récemment publié (avril 2022), reprenant les bases de la génétique ainsi que les diverses indications pour réaliser un test génétique dans le cadre des canalopathies, des cardiomyopathies, des morts subites, des pathologies cardiaques congénitales, des maladies coronariennes et de l'insuffisance cardiaque.1 Grâce au développement des techniques de séquençage, le nombre de gènes pouvant être séquencés a grandement augmenté, et les résultats génétiques sont disponibles de plus en plus rapidement. En conséquence, et grâce à une interprétation plus standardisée des variants génétiques, les connaissances sur l'hérédité des pathologies cardiaques ainsi que sur les mécanismes physiopathologiques sous-jacents ont grandement évolué.1
L'hérédité des maladies cardiaques : le modèle mendélien et le modèle oligo-/ polygénique
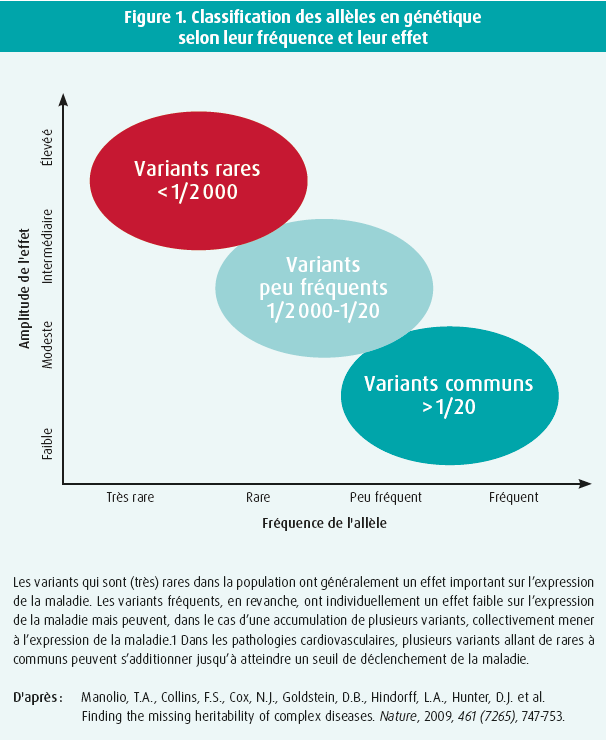
On postulait auparavant que les maladies cardiaques rares, comme les autres maladies génétiques, étaient héritées en respectant les lois de Mendel, c.-à-d. que leur hérédité pouvait s'expliquer par la transmission d'un seul variant génétique rare. Ce concept a ensuite évolué vers un spectre plus large allant d'une hérédité mendélienne monogénique à une hérédité oligogénique ou complexe polygénique, dans laquelle l'interaction entre plusieurs variants communs module l'expressivité de la pathologie (figure 1). Au sein de ce spectre, différentes combinaisons sont possibles, allant de variants rares à fréquents avec un effet respectivement large ou faible, pouvant conduire au développement de la maladie de manière isolée ou en combinaison variable selon la pathologie (figure 1). Ce modèle pourrait expliquer la pénétrance incomplète et la variabilité phénotypique au sein des familles. En outre, cette théorie expliquerait le faible rendement des analyses génétiques, axées sur l'hérédité mendélienne, dans certaines pathologies.2 à l'heure actuelle, d'autres outils génétiques sont en développement, afin de mieux comprendre la variabilité d'expression et de pénétrance des maladies cardiovasculaires au sein d'une même famille. Il s'agit notamment des scores polygéniques de risque qui, à ce jour, ont surtout été utilisés en recherche, mais auxquels on a de plus en plus recours dans les essais cliniques, et dont le rôle clinique reste à définir.1
Conséquences des tests génétiques pour le patient et les membres de sa famille
Les tests génétiques doivent tout d'abord être réalisés chez le patient pour lequel un diagnostic clinique a été posé. La mise en évidence d'un variant pathogène permet alors de confirmer ce diagnostic clinique et d'adapter le suivi. Une fois le variant pathogène identifié, celui-ci pourra servir d'outil de stratification du risque au sein de la famille, en réalisant ce que l'on appelle une « analyse en cascade », en commençant par les proches au premier degré du patient. De cette manière, les porteurs du variant pathogène peuvent bénéficier d'un suivi adapté, et les membres non porteurs du variant familial peuvent être rassurés. Dans certains cas, la première présentation d'une pathologie cardiaque héréditaire est une arythmie maligne, pouvant parfois mener à une mort subite. L'autopsie moléculaire, consistant en une analyse génétique post mortem, a une place grandissante dans ces situations. En effet, l'identification d'un potentiel facteur génétique causal permet de préciser l'étiologie de la mort subite, mais aussi d'identifier précocement les proches du défunt à risque, afin d'appliquer, le cas échéant, des traitements préventifs. Certaines études ont démontré un rendement atteignant jusqu'à 35 % de diagnostic moléculaire à l'aide des nouvelles techniques de séquençage.4
Les thérapies géniques dans le cadre de la cardiogénétique
Outre l'objectif diagnostique des tests génétiques pour les patients et les membres de leur famille, la découverte d'un variant génétique peut également avoir une valeur pronostique et thérapeutique, en fonction de la pathologie. L'identification de variants pathogènes dans diverses pathologies cardiovasculaires favorise le développement des connaissances sur les mécanismes physio pathologiques sous-jacents, ce qui permet également de développer des traitements spécifiques. Par exemple, dans le cadre des cardiomyopathies hypertrophiques, causées par des variants pathogènes dans l'un des gènes du sarcomère, le métabolisme énergétique des cardiomyocytes est altéré secondairement à un état de tension des ponts entre l'actine et la myosine.5 Le mavacamten, un inhibiteur de la myosine ATPase cardiaque, a été développé dans ce contexte et testé dans l'essai clinique EXPLORER-HCM, dans lequel les auteurs ont pu démontrer une amélioration significative des symptômes cliniques chez les patients atteints de cardiomyopathie hypertrophique obstructive.6
D'autre part, les thérapies géniques, utilisant les ARN interférents (siRNA), ont été développées principalement dans le cadre de l'hypercholestérolémie.7 Par exemple, l'inclisiran, un siRNA qui cible le foie pour inhiber la synthèse de la proprotéine convertase subtilisine/kexine de type 9 (PCSK9), entraîne une réduction du LDL d'environ 50 %.8 Une autre forme de thérapie génique actuellement explorée est la modification du génome via la technique du CRISPR/Cas9, que l'on peut qualifier de « ciseaux moléculaires ». Cette protéine permet de cibler un endroit précis d'un gène pour y induire une cassure de l'ADN. Un système de réparation se met ensuite en place, dont deux types existent : l'un permettant de corriger précisément le variant pathogène, et l'autre, de remettre l'ADN bout à bout mais avec des erreurs, entraînant une modification du cadre de lecture du gène et empêchant la production d'une protéine d'intérêt.9 La technique CRISPR/ Cas9 est actuellement étudiée pour être appliquée dans le cadre de l'amylose. La molécule NTLA-2001 utilisée à cette fin contient un système d'administration, composé de nanoparticules lipidiques qui cible le foie, et un ARN guide unique (sgRNA) qui cible la transthyrétine humaine (TTR) dans les hépatocytes. De cette manière, la production, et donc l'accumulation, de TTR peuvent être réduites.10 Cependant, il est actuellement difficile d'appliquer la technique CRISPR/ Cas9 aux cardiomyocytes, car il n'existe pas encore de système d'administration approprié pour cibler ce type cellulaire.
Obtenir des informations à partir du puzzle qu'est le génome : le défi clinique de la (ré)interprétation des variants
Bart Loeys - Centre de génétique médicale, UZ Antwerp
Au cours des dernières années, on a observé une amélioration exponentielle de la technologie des tests génétiques. Grâce à cette évolution, de plus en plus de gènes peuvent être analysés en même temps, à moindre coût et plus rapidement. Ceci mène à l'identification de plus de variants, qu'il faut alors pouvoir interpréter. Lors de la découverte d'un variant génétique, il est extrêmement important d'évaluer correctement sa pathogénicité. Une interprétation systématique de celle-ci n'est actuellement possible que pour les régions codantes du génome. Il est cependant important de noter que la classification des variants peut évoluer au cours du temps et varier selon les connaissances sur la maladie considérée. Il faut donc parfois réévaluer la pathogénicité des variants identifiés.
évolution de la nomenclature
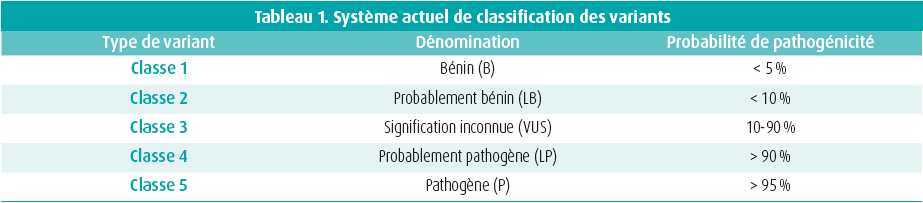
Les variants génétiques ont été nommés différemment au cours des dernières années. Par le passé, les dénominations « mutation » et « polymorphisme » étaient utilisées et interprétées comme étant respectivement à l'origine de maladies ou de nature inoffensive. Récemment, cette nomenclature a été réformée pour adopter un système plus neutre et plus nuancé. Toute modification du génome est décrite comme un « variant ». Une dénomination plus détaillée du variant donne un aperçu de la probabilité de pathogénicité. Les variants sont décrits comme « bénins (B) », « probablement bénins (LB) », « variants de signification inconnue (VUS) », « probablement pathogènes (LP) » et « pathogènes (P) ». Les variants LB et LP ont respectivement 10 et 90 % de chances d'être pathogènes (tableau 1).11
Classification des variants
Les variants sont évalués d'un point de vue moléculaire et clinique, pour déterminer dans quelle mesure ils peuvent expliquer le tableau clinique d'un patient. Il est important de noter que, comme pour de nombreux aspects de la médecine, il s'agit d'un processus dynamique qui évolue au fil des ans. L'interprétation moléculaire commence généralement par l'évaluation des données précédemment publiées concernant le gène ou le variant spécifique. La base de données la plus fréquemment utilisée pour recueillir les données génotypiques et phénotypiques est ClinVar.12 Ensuite, on étudie la fréquence du variant spécifique dans des populations témoins. La base de données la plus fréquemment utilisée à cette fin est gnomAD, qui contient 250 000 allèles provenant d'individus (présumés) sains.13 Les variants plus fréquents dans gnomAD sont moins susceptibles d'être pathogènes, et vice versa. Cependant, la fréquence attendue dépend de la pathologie spécifique étudiée, ainsi que de l'origine ethnique. Les effets attendus du variant au niveau de la protéine influencent également la probabilité de pathogénicité : 1) changement de la propriété de l'acide aminé (polarité et charge) ; 2) conservation de l'acide aminé : le degré de conservation de l'acide aminé entre différentes espèces donne une idée de l'importance de l'acide aminé spécifique ; 3) domaine fonctionnel de la protéine dans lequel le variant est situé. Enfin, dans un nombre limité de cas, on a accès à des données fonctionnelles, vérifiant la fonction de la protéine altérée. L'interprétation clinique du variant doit être associée à l'interprétation moléculaire, en y ajoutant des données cliniques, des antécédents familiaux et des données de ségrégation.
Les informations susmentionnées sont compilées dans un système de classification standardisé, dans lequel plusieurs degrés de validité sont attribués à chaque critère. L'interprétation finale classe le variant dans l'une des cinq classes précédemment citées.11 Il est important que la classification des variants soit effectuée par une équipe multidisciplinaire comprenant des experts moléculaires et cliniques.
Déclaration des variants
Les variants de classe 1 et 2 (B et LB) ne sont généralement pas déclarés ni utilisés dans le cadre clinique. Les variants de classe 4 et 5 (L et LP) sont toujours signalés et sont également utilisés pour des tests prédictifs chez les membres de la famille. Dans le cas d'un variant de classe 4, il peut également être utile de réaliser une analyse supplémentaire de ségrégation, afin de vérifier sa signification et d'élever sa classification à la classe 5. Dans le cas d'un variant de classe 3 (VUS), l'incertitude persiste quant à la prise en charge optimale. à l'UZ Antwerp, les variants de ce type sont signalés et divisés en deux groupes. En cas de « VUS froid » (pathogène moins probable), la ségrégation est effectuée uniquement chez les parents au premier degré, et principalement pour optimiser l'interprétation du variant ; le suivi clinique est assuré indépendamment de ce résultat. En cas de « VUS chaud » (pathogène possible, mais avec des données encore insuffisantes), la ségrégation génétique chez les parents au premier degré est assurée, et le résultat détermine la fréquence du suivi clinique.
Reclassification des variants
Comme indiqué précédemment, la cardiogénétique est une discipline en plein essor. Ainsi, avec le temps, on observe une évolution significative des connaissances concernant les variants génétiques. Cela se traduit p.ex. par une mise à jour des lignes directrices générales, par l'ajout de lignes directrices de classification spécifiques aux gènes ou par des informations moléculaires/cliniques supplémentaires sur des variants spécifiques.14 Il est important de continuer à reclasser les variants précédemment identifiés, comme décrit ci-dessus, en fonction des connaissances actuelles. En effet, une telle reclassification peut avoir un impact significatif pour le patient et sa famille. Un variant (probablement) pathogène peut, après réévaluation, être décrit comme « VUS » ou « (probablement) bénin », ou vice versa, ce qui peut entraîner un changement significatif dans la prise en charge du patient et de sa famille.15
Meilleure présentation d'abstract : le co-transporteur 1 du sodium myo-inositol affecte l'hypertrophie cardiaque dans les coeurs de souris surchargés en pression
Alice Marino - Centre de Recherche Cardiovasculaire, UC Louvain
L'insuffisance cardiaque est une maladie dont la morbidité et la mortalité sont connues pour être élevées.16 Plusieurs changements sont observés dans le cadre de l'insuffisance cardiaque : hypertrophie, dysfonctionnement endothélial, fibrose, inflammation et modifications dans les métabolites.17, 18, 19 L'un des métabolites qui augmentent dans l'insuffisance cardiaque avancée est le myo-inositol.17 Celui-ci se déplace, via SMIT1 (co-transporteur 1 du sodium myo-inositol), dans les cellules, où le myo-inositol est ensuite métabolisé. SMIT1 joue également un rôle important dans la glucotoxicité myocardique observée en cas d'hyperglycémie.20
Pour déterminer plus précisément le rôle de SMIT1, une constriction aortique transversale a été réalisée chirurgicalement sur des souris Smit1-knockout (KO) et des souris wild type (WT). Les souris KO Smit1 présentent une fonction cardiaque mieux préservée et une hypertrophie myocardique échocardiographiquement moins prononcée après cette procédure. Histologiquement, une quantité moindre d'hypertrophie et de fibrose myocardique est confirmée par rapport aux souris WT. Les données de séquençage de l'ARN montrent une régulation négative des gènes liés à l'hypertrophie et à la fibrose chez les souris KO. Lorsque l'on administre de la phényléphrine, qui induit également une charge hémodynamique accrue, on observe une hypertrophie myocardique moins prononcée chez les souris KO que chez les souris WT. Ces résultats montrent que SMIT1 serait potentiellement une cible intéressante pour prévenir l'insuffisance cardiaque.
Références
- Wilde, A.A.M., Semsarian, C., Marquez, M.F., Shamloo, A.S., Ackerman, M.J., Ashley, E.A. et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace, 2022, 24 (8), 1307-1367.
- Walsh, R., Tadros, R., Bezzina, C.R. When genetic burden reaches threshold. Eur Heart J, 2020, 41 (39), 3849-3855.
- Manolio, T.A., Collins, F.S., Cox, N.J., Goldstein, D.B., Hindorff, L.A., Hunter, D.J. et al. Finding the missing heritability of complex diseases. Nature, 2009, 461 (7265), 747-753.
- Castiglione, V., Modena, M., Aimo, A., Chiti, E., Botto, N., Vittorini, S. et al. Molecular Autopsy of Sudden Cardiac Death in the Genomics Era. Diagnostics (Basel), 2021, 11 (8), 1378.
- Trivedi, D.V., Adhikari, A.S., Sarkar, S.S., Ruppel, K.M., Spudich, J.A. Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev, 2018, 10 (1), 27-48.
- Olivotto, I., Oreziak, A., Barriales-Villa, R., Abraham, T.P., Masri, A., Garcia-Pavia, P. et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet, 2020, 396 (10253), 759-769.
- Katzmann, J.L., Packard, C.J., Chapman, M.J., Katzmann, I., Laufs, U. Targeting RNA With Antisense Oligonucleotides and Small Interfering RNA: JACC State-of-the-Art Review. J Am Coll Cardiol, 2020, 76 (5), 563-579.
- Ray, K.K., Wright, R.S., Kallend, D., Koenig, W., Leiter, L.A., Raal, F.J. et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med, 2020, 382 (16), 1507-1519.
- Vermersch, E., Jouve, C., Hulot, J.S. CRISPR/Cas9 gene-editing strategies in cardiovascular cells. Cardiovasc Res, 2020, 116 (5), 894-907.
- Gillmore, J.D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M.L. et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med, 2021, 385 (6), 493-502.
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17 (5), 405-424.
- Landrum, M.J., Lee, J.M., Benson, M., Brown, G.R., Chao, C., Chitipiralla, S. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res, 2018, 46 (D1), D1062-D7.
- Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 2020, 581 (7809), 434-443.
- Muiño-Mosquera, L., Steijns, F., Audenaert, T., Meerschaut, I., De Paepe, A., Steyaert, W. et al. Tailoring the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Guidelines for the Interpretation of Sequenced Variants in the FBN1 Gene for Marfan Syndrome: Proposal for a Diseaseand Gene-Specific Guideline. Circ Genom Precis Med, 2018, 11 (6), e002039.
- VanDyke, R.E., Hashimoto, S., Morales, A., Pyatt, R.E., Sturm, A.C. Impact of variant reclassification in the clinical setting of cardiovascular genetics. J Genet Couns, 2021, 30 (2), 503-512.
- Ziaeian, B., Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat Rev Cardiol, 2016, 13 (6), 368-378.
- Deidda, M., Piras, C., Dessalvi, C.C., Locci, E., Barberini, L., Torri, F. et al. Metabolomic approach to profile functional and metabolic changes in heart failure. J Transl Med, 2015, 13, 297.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc Res, 2021, 117 (6), 1450-1488.
- Murphy, S.P., Kakkar, R., McCarthy, C.P., Januzzi, J.L.Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J Am Coll Cardiol, 2020, 75 (11), 1324-1340.
- Van Steenbergen, A., Balteau, M., Ginion, A., Ferte, L., Battault, S., Ravenstein, C.M. et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci Rep, 2017, 7, 41166.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.