BSC-congresverslag - sessie 24
Op het jaarlijkse congres van de Belgische Vereniging voor Cardiologie (BSC) organiseerde de Belgian Council on Cardiogenomics een sessie rond de rol van de genetica binnen de cardiologie. Tijdens die sessie lichtten de verschillende presentatoren de implicaties en het belang van genetische tests binnen de cardiologie toe. Dit voornamelijk om de klinische diagnose te bevestigen, de prognose beter in te schatten en in sommige gevallen ook de behandeling bij te sturen. Daarnaast kan een genetische test familieleden die een risico hebben om dezelfde aandoening te ontwikkelen vroegtijdig identificeren. De verschillende resultaten die bij een genetische test kunnen worden verkregen, werden besproken. Ook het proces voor de interpretatie en classificatie van een genetische variant werd toegelicht. Genetische counseling speelt een centrale rol in dat proces en biedt een kader om de limieten van de beschikbare testen te duiden, de implicaties van de resultaten (voor de patiënt en zijn familieleden) uit te leggen en de psychologische impact van de diagnose in te schatten.
Genetische testen bij cardiale aandoeningen: een stand van zaken in een snel evoluerend domein
James Ware - Imperial College, Londen, VK
Cardiogenetica is een discipline in volle ontwikkeling. In april 2022 werd een expertconsensusdocument gepubliceerd waarin de grondbeginselen van de genetica en de verschillende indicaties voor genetische tests bij channelopathieën, cardiomyopathieën, plotse dood, congenitale hartafwijkingen, coronaire hartziekten en hartfalen werden uiteengezet.1 Dankzij de ontwikkeling van sequencingtechnieken is het aantal analyseerbare genen, en de snelheid waarmee genetische resultaten worden bekomen enorm toegenomen. Als gevolg daarvan en dankzij een meer gestandaardiseerde manier om genetische varianten te interpreteren, is de kennis over de erfelijkheid en de onderliggende pathofysiologische mechanismen van hartaandoeningen sterk geëvolueerd.1
Overerving van cardiale aandoeningen: het mendeliaanse versus het oligopolygene model
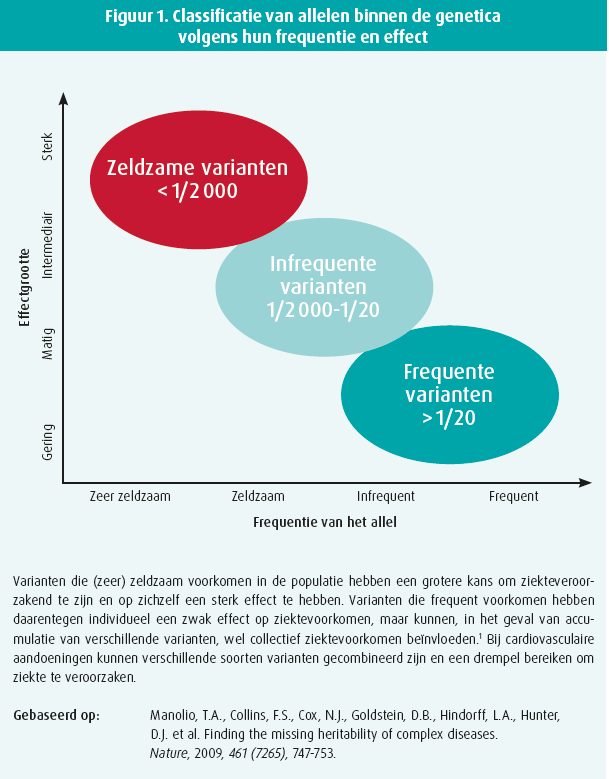
Oorspronkelijk werd verondersteld dat zeldzame hartziekten, net zoals andere genetische aandoeningen, louter mendeliaans - dus door de overerving van één zeldzame genetische variant - werden overgeërfd. Dat concept ontwikkelde zich tot een breder spectrum gaande van een monogene mendeliaanse tot een oligogene overerving of een complexe polygene overerving, waarbij de interactie van verschillende frequentere varianten de expressie van de ziekte moduleren (figuur 1). Binnen dit spectrum kunnen verschillende combinaties, van zeldzame tot frequente varianten met respectievelijk een groot of klein effect, leiden tot de ontwikkeling van de ziekte (figuur 1). Dit model zou de onvolledige penetrantie en fenotypische variabiliteit binnen families kunnen verklaren. Daarnaast zou het een verklaring bieden voor het lage rendement van genetische testen, gefocust op mendeliaanse overerving, bij bepaalde aandoeningen.2 Tot op heden worden bijkomende genetische tools ontwikkeld om de variabele expressie en penetrantie van cardiovasculaire aandoeningen binnen dezelfde familie beter in te schatten. Daarbij gaat het om polygene risicoscores die tot dusver vooral werden gebruikt in onderzoek, maar nu ook hun weg vinden in de klinische setting.1 De rol ervan in deze context moet dus nog worden bepaald.
Consequenties van genetische testing voor patiënt en familieleden
Genetische testing begint steeds bij de patiënt waarbij een klinische diagnose werd gesteld. De identificatie van een pathogene variant maakt het mogelijk om de klinische diagnose te bevestigen en eventueel bijkomende geassocieerde problemen vroegtijdig op te sporen. Eens een pathogene variant binnen een familie wordt geïdentificeerd, kan men een risicovoorspelling binnen de familie uitvoeren door middel van een cascadeanalyse, beginnend bij de eerstegraadsfamilieleden. Op die manier kan men de dragers van de pathogene variant van de nodige langetermijnsopvolging voorzien en kunnen de familieleden die de pathogene variant niet dragen, worden gerustgesteld. In sommige gevallen is de eerste presentatie van een overerfbare cardiale aandoening een maligne aritmie die kan leiden tot plotse dood. Moleculaire autopsie, bestaande uit een post mortem genetische analyse, neemt in deze situaties een belangrijke plaats in. Zo'n analyse maakt het mogelijk om een potentiële genetische oorzakelijke factor op te sporen en zo een verklaring te bieden voor de plotse dood. De identificatie van een pathogene variant laat in dit geval toe familieleden van de overledene met risico op plotse dood vroegtijdig te identificeren en op die manier preventieve behandelingen, waar nodig, toe te passen. Het rendement van moleculaire diagnose met nieuwe sequencingtechnieken kan tot 35 % bedragen.4
Genetische therapieën binnen de cardiogenetica
Naast het diagnostische doel van genetische tests voor zowel de patiënt als zijn of haar familieleden, kan de ontdekking van een pathogene genetische variant, afhankelijk van de specifieke ziekte, ook een prognostische en therapeutische waarde hebben. Door de identificatie van pathogene varianten in verschillende harten vaatziekten neemt de kennis over de onderliggende pathofysiologische mechanismen toe, wat toelaat om ook specifieke behandelingen te ontwikkelen. Bijvoorbeeld bij hypertrofische cardiomyopathie, veroorzaakt door pathogene varianten in een van de sarcomeergenen, verandert het energiemetabolisme van de cardiomyocyten ten gevolge van een spanningstoestand ter hoogte van de overbruggingen tussen actine en myosine.5 Mavacamten, een inhibitor van het cardiale myosine ATP-ase, werd ontwikkeld in deze context en werd in de klinische EXPLORER-HCM-trial getest. In die trial hebben de onderzoekers een significante verbetering van de klinische symptomen kunnen aantonen bij patiënten met obstructieve hypertrofe cardiomyopathie.6
Gentherapieën, waarbij gebruik wordt gemaakt van interfererende RNA's (siRNA's), werden voornamelijk ontwikkeld voor hypercholesterolemie.7 Bijvoorbeeld inclisiran, een siRNA die zich op de lever richt om de synthese van het pro-eiwit convertase subtilisin-kexin type 9 (PCSK9) te inhiberen, leidt tot een reductie van ongeveer 50 % LDL in het bloed.8 Een andere specifieke gentherapie die momenteel wordt geëxploreerd, is de rechtstreekse wijziging van een gen via de CRISPR/Cas9-techniek, wat kan worden omschreven als een moleculaire schaar. Die techniek laat toe om een specifiek punt in een gen te knippen. Vervolgens wordt een reparatiesysteem in werking gesteld, waarbij er twee mogelijkheden zijn: 1) de pathogene variant kan precies worden gecorrigeerd, 2) het DNA wordt weer in elkaar gezet, maar met een fout, waardoor het leesraam van het gen verandert en de productie van het gewenste eiwit wordt verhinderd.9 De CRISPR/Cas9-techniek wordt momenteel bestudeerd voor toepassing bij amyloïdose. De NTLA-2001-molecule die hierbij wordt gebruikt, bevat een afleveringssysteem met een lipide nanopartikel dat de lever target en een single guide RNA (sgRNA) die zich richt op het menselijke transthyretine (TTR) in hepatocyten. Op die manier kan de productie en dus ook accumulatie van TTR worden verminderd.10 Momenteel is het echter moeilijk om de CRISPR/Cas9-techniek toe te passen in cardiomyocyten, aangezien er nog geen goed afleveringssysteem bestaat dat zich richt op dit celtype.
Informatie verkrijgen uit een chaotische achtergrond: de klinische uitdaging van de (her)interpretatie van varianten
Bart Loeys - Centrum voor Medische Genetica, UZ Antwerpen
De voorbije jaren verbeterde de technologie van genetische testen exponentieel. Die evolutie zorgt ervoor dat men steeds goedkoper, sneller en meer genen tegelijkertijd kan analyseren. Daardoor kunnen er ook meer varianten worden gevonden. Wanneer een genetische variant gevonden wordt, is het uiterst belangrijk de pathogeniciteit correct in te schatten. Een systematische interpretatie hiervan is tot op heden enkel mogelijk voor de coderende regio's van het genoom. Belangrijk is dat de classificatie van varianten evolueert doorheen de tijd en varieert in functie van de kennis over de specifieke aandoening. Het is soms dus nodig om de pathogeniciteit van vastgestelde varianten te herevalueren.
Evolutie van de nomenclatuur
Veranderingen in het genoom werden de voorbije jaren op verschillende manieren benoemd. In het verleden werd frequent gesproken over mutaties en polymorfismen wanneer deze werden geïnterpreteerd als respectievelijk ziekteveroorzakend of onschuldig. Recent werd deze nomenclatuur hervormd naar een neutraler en genuanceerder systeem. Iedere aanpassing in het genoom wordt nu beschreven als een variant. Een meer gedetailleerde benaming van de variant geeft een inzicht op de kans op pathogeniciteit. Varianten worden beschreven als benigne (B), waarschijnlijk benigne (likely benign, LB), variant van ongekende betekenis (variant of unknown significance, VUS), waarschijnlijk pathogeen (likely pathogenic, LP) en pathogeen (P). Hierbij hebben LB en LP-varianten respectievelijk een kans van 10 % en 90 % om daadwerkelijk pathogeen te zijn (tabel 1).11

Variantclassificatie
Varianten worden zowel op moleculair als klinisch vlak geëvalueerd op de mate waarin zij het klinische beeld bij een patiënt kunnen verklaren. Belangrijk is dat dit - zoals vele aspecten binnen de geneeskunde - een dynamisch proces is, evolutief door de jaren heen. De moleculaire interpretatie begint doorgaans met de evaluatie van eerder gepubliceerde data rond het specifieke gen of de variant. De meest frequent gebruikte database die geno- en fenotypische data verzamelt is ClinVar.12 Nadien wordt de frequentie van de specifieke variant in controlepopulaties nagegaan. Hiervoor wordt het vaakst gebruik gemaakt van de database gnomAD, die 250 000 allelen bevat van (vermoedelijk) gezonde individuen.13 Frequentere varianten in gnomAD zijn minder waarschijnlijk pathogeen en omgekeerd geldt dat varianten die minder vaak voorkomen of afwezig zijn in gnomAD een verhoogde kans hebben om pathogeen te zijn. De verwachte frequentie is echter afhankelijk van de specifiek onderzochte aandoening, en ook van de etniciteit. Ook de verwachte effecten van de variant op eiwitniveau beïnvloeden de waarschijnlijkheid op pathogeniciteit: 1) verandering van de eigenschap van het aminozuur (polariteit en lading), 2) conservatie van het aminozuur: de mate waarin het aminozuur bewaard bleef over verschillende diersoorten heen geeft een indruk van het belang van het specifieke aminozuur, en 3) het functionele domein van het eiwit waarin de variant zich bevindt. Tot slot heeft men in een beperkt aantal gevallen toegang tot functionele data, waarbij men de functie van het veranderde eiwit nagaat. In combinatie met de moleculaire interpretatie moet ook een klinische interpretatie van de variant gebeuren. Dat vindt op een multidisciplinaire manier plaats en voegt klinische data, familiale voorgeschiedenis en segregatiedata toe aan de moleculaire interpretatie.
Deze informatie wordt samengebracht in een gestandaardiseerd classificatiesysteem. Verschillende graden van validiteit worden toegekend aan criteria, waarbij de uiteindelijke opsomming de variant indeelt in een van de vijf klassen.11 Belangrijk hierbij is dat variantclassificatie gebeurt in een multidisciplinair team waarbij zowel moleculaire als klinische experten aanwezig zijn.
Rapportering van varianten
Klasse 1- en 2-varianten (B en LB) worden meestal niet gerapporteerd en worden niet gebruikt in de klinische setting. Klasse 4- en 5-varianten (LP en P) worden steeds gerapporteerd en worden ook gebruikt voor voorspellende testen bij familieleden. In het geval van een klasse 4-variant kan het ook nuttig zijn een verdere segregatieanalyse uit te voeren om de significantie ervan na te gaan en de classificatie van de variant naar een klasse 5 te verhogen. In het geval van een klasse 3-variant (VUS) bestaat er nog onduidelijkheid rond het optimale beleid. In het UZA worden deze gerapporteerd en verder opgedeeld in twee groepen. Bij een 'koude VUS' (minder waarschijnlijk pathogeen) wordt segregatie enkel uitgevoerd op eerstegraadsverwanten en dat voornamelijk om de interpretatie van de variant te optimaliseren; klinische follow-up wordt onafhankelijk van dit resultaat voorzien. Bij een 'warme VUS' (mogelijks pathogeen, maar met nog onvoldoende gegevens) wordt genetische segregatie van eerstegraadsverwanten voorzien en bepaalt het resultaat de frequentie van klinische follow-up.
Variant-herclassificatie
Zoals vermeld is cardiogenetica een discipline in volle ontwikkeling. Hierbij wordt met de tijd een aanzienlijke evolutie in de kennis over genetische varianten gezien. Dit wordt bijvoorbeeld gezien onder de vorm van een update van de algemene richtlijnen, toevoeging van genspecifieke classificatierichtlijnen of bijkomende moleculaire/klinische informatie over specifieke varianten.14 Belangrijk hierbij is om de classificatie van eerder bepaalde varianten, zoals deze hierboven werd beschreven, te blijven toetsen aan de huidige kennis. Een dergelijke herclassificatie kan namelijk een belangrijke impact hebben op de patiënt en zijn familie, waarbij een (waarschijnlijk) pathogene variant toch als VUS of (waarschijnlijk) benigne kan worden omschreven of omgekeerd. Dit kan een belangrijke verandering teweegbrengen in het medische beleid.15
Beste abstract presentatie: de natrium myo-inositol co-transporter 1 beïnvloedt cardiale hypertrofie in druk-overbelaste muizenharten
Alice Marino - Centrum voor Cardiovasculair Onderzoek, UC Louvain
Hartfalen is een aandoening met een gekende hoge morbiditeit en mortaliteit.16 Bij het instellen van hartfalen worden verscheidene veranderingen gezien: hypertrofie, endotheliale disfunctie, fibrose, inflammatie en veranderingen in metabolieten.17, 18, 19 Een van de metabolieten die stijgt bij vergevorderd hartfalen is myo-inositol.17 Deze verplaatst zich intracellulair via SMIT1, de natrium myo-inositol co-transporter 1, waar myo-inositol verder wordt gemetaboliseerd. SMIT1 speelt verder ook een belangrijke rol bij de myocardiale glucotoxiciteit die wordt gezien bij hyperglykemie.20
Om de rol van SMIT1 verder te bepalen werd bij Smit1 knock-out (KO)- en wildtype (WT)-muizen chirurgisch een dwarse aortaconstrictie aangebracht. Smit1 KO-muizen vertonen na deze ingreep echocardiografisch een beter bewaarde hartfunctie en minder uitgesproken myocardiale hypertrofie. Histologisch wordt een lagere hoeveelheid myocardiale hypertrofie en fibrose bevestigd ten opzichte van WT-muizen. RNA-sequencing data tonen een down-regulatie van de genen gerelateerd aan hypertrofie en fibrose in KO-muizen. Bij toediening van fenylefrine, wat ook een verhoogde hemodynamische load in de hand werkt, wordt bij KO-muizen een minder uitgesproken myocardiale hypertrofie gezien ten opzichte van WT-muizen. Deze bevindingen tonen aan dat SMIT1 mogelijk een interessant target zou kunnen vormen om hartfalen te voorkomen.
Referenties
- Wilde, A.A.M., Semsarian, C., Marquez, M.F., Shamloo, A.S., Ackerman, M.J., Ashley, E.A. et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace, 2022, 24 (8), 1307-1367.
- Walsh, R., Tadros, R., Bezzina, C.R. When genetic burden reaches threshold. Eur Heart J, 2020, 41 (39), 3849-3855.
- Manolio, T.A., Collins, F.S., Cox, N.J., Goldstein, D.B., Hindorff, L.A., Hunter, D.J. et al. Finding the missing heritability of complex diseases. Nature, 2009, 461 (7265), 747-753.
- Castiglione, V., Modena, M., Aimo, A., Chiti, E., Botto, N., Vittorini, S. et al. Molecular Autopsy of Sudden Cardiac Death in the Genomics Era. Diagnostics (Basel), 2021, 11 (8), 1378.
- Trivedi, D.V., Adhikari, A.S., Sarkar, S.S., Ruppel, K.M., Spudich, J.A. Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev, 2018, 10 (1), 27-48.
- Olivotto, I., Oreziak, A., Barriales-Villa, R., Abraham, T.P., Masri, A., Garcia-Pavia, P. et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, doubleblind, placebocontrolled, phase 3 trial. The Lancet, 2020, 396 (10253), 759-769.
- Katzmann, J.L., Packard, C.J., Chapman, M.J., Katzmann, I., Laufs, U. Targeting RNA With Antisense Oligonucleotides and Small Interfering RNA: JACC State-of-the-Art Review. J Am Coll Cardiol, 2020, 76 (5), 563-579.
- Ray, K.K., Wright, R.S., Kallend, D., Koenig, W., Leiter, L.A., Raal, F.J. et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med, 2020, 382 (16), 1507-1519.
- Vermersch, E., Jouve, C., Hulot, J.S. CRISPR/Cas9 gene-editing strategies in cardiovascular cells. Cardiovasc Res, 2020, 116 (5), 894-907.
- Gillmore, J.D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M.L. et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med, 2021, 385 (6), 493-502.
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015, 17 (5), 405-424.
- Landrum, M.J., Lee, J.M., Benson, M., Brown, G.R., Chao, C., Chitipiralla, S. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res, 2018, 46 (D1), D1062D7.
- Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 2020, 581 (7809), 434-443.
- Muiño-Mosquera, L., Steijns, F., Audenaert, T., Meerschaut, I., De Paepe, A., Steyaert, W. et al. Tailoring the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Guidelines for the Interpretation of Sequenced Variants in the FBN1 Gene for Marfan Syndrome: Proposal for a Diseaseand Gene-Specific Guideline. Circ Genom Precis Med, 2018, 11 (6), e002039.
- VanDyke, R.E., Hashimoto, S., Morales, A., Pyatt, R.E., Sturm, A.C. Impact of variant reclassification in the clinical setting of cardiovascular genetics. J Genet Couns, 2021, 30 (2), 503-512.
- Ziaeian, B., Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat Rev Cardiol, 2016, 13 (6), 368-378.
- Deidda, M., Piras, C., Dessalvi, C.C., Locci, E., Barberini, L., Torri, F. et al. Metabolomic approach to profile functional and metabolic changes in heart failure. J Transl Med, 2015, 13, 297.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc Res, 2021, 117 (6), 1450-1488.
- Murphy, S.P., Kakkar, R., McCarthy, C.P., Januzzi, J.L.Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J Am Coll Cardiol, 2020, v, 1324-1340.
- Van Steenbergen, A., Balteau, M., Ginion, A., Ferte, L., Battault, S., Ravenstein, C.M. et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci Rep, 2017, 7, 41166.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.