Compte rendu du congrès de l'ESC
Comme le veut la tradition annuelle, ces vacances d'été se sont une nouvelle fois clôturées sur le congrès de la European Society of Cardiology (ESC), qui s'est tenu cette année à Amsterdam, du 25 au 28 août 2023. Comme toujours, quatre nouvelles recommandations y ont été présentées, dont les toutes premières recommandations de l'ESC pour la prise en charge des cardiomyopathies.1 Outre une actualisation approfondie des recommandations (après près de dix ans) sur la cardiomyopathie hypertrophique2, ce document contient des recommandations totalement inédites sur d'autres phénotypes. La session s'est déroulée le dimanche 27 août, sous la houlette des deux présidents : Elena Arbelo (Barcelone, Espagne) et Juan Pablo Kaski (Londres, Royaume-Uni).
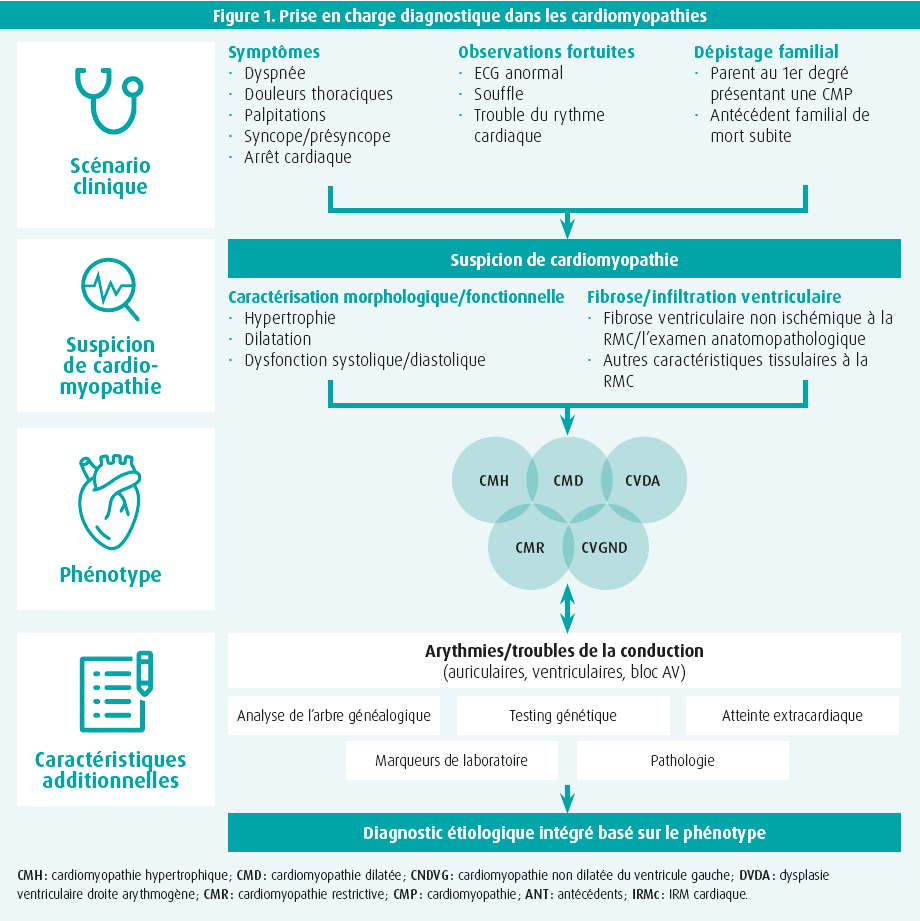
Quiconque traite régulièrement, dans sa pratique clinique, des patients atteints de cardiomyopathie sait que chaque prise en charge débute par une question diagnostique, en fonction du phénotype au moment de la présentation. Il est recommandé de décrire le phénotype sur la base de quelques caractéristiques morphologiques, telles qu'une hypertrophie et/ou une dilatation ventriculaire droite/gauche, ainsi que de la présence éventuelle d'une cicatrice (non ischémique) à l'IRM. Pour le reste, des caractéristiques fonctionnelles sont également utilisées pour définir le type de cardiomyopathie, comme une dysfonction systolique et/ou diastolique de l'un ou des deux ventricules. Parallèlement à la cardiomyopathie hypertrophique (CMH), dilatée (CMD) et restrictive (CMR), l'usage du terme 'dysplasie ventriculaire droite arythmogène' (DVDA) est à nouveau conseillé dans cette recommandation.1, 3 Les patients qui ont une cardiomyopathie arythmogène gauche sont actuellement repris sous le terme générique 'cardiomyopathie non dilatée du ventricule gauche' (CNDVG). Ce nouveau phénotype de CNDVG est défini par les auteurs comme 1) la présence d'une fibrose non ischémique ou d'une maladie infiltrante du ventricule gauche, indépendamment de troubles de la contractilité segmentaire ou globale ou 2) une hypokinésie ventriculaire gauche isolée en l'absence de fibrose.4 En parallèle, le phénotype de non-compaction du ventricule gauche n'est plus considéré comme une cardiomyopathie en soi, mais on parle plutôt d'hypertrabéculation comme d'un phénomène qui peut se produire de manière isolée ou en association avec une hypertrophie, une dilatation et/ou une dysfonction systolique ventriculaire gauche. Les recommandations actuelles ne considèrent plus non plus le syndrome de takotsubo - étant donné la nature passagère du phénomène - comme une cardiomyopathie.1
Outre une description phénotypique détaillée de chaque cardiomyopathie, un diagnostic étiologique correct est crucial pour déterminer la suite de la prise en charge, bien qu'il puisse s'agir d'une véritable gageure en pratique clinique. C'est justement la raison pour laquelle une grande attention est accordée, tant par les auteurs de la recommandation que par Elena Biagini (Bologne, Italie) dans cette session, au workflow diagnostique (figure 1). Comme toujours, la première étape est une évaluation clinique approfondie, consacrant une attention spécifique à la symptomatologie, aux antécédents familiaux et aux signes cliniques susceptibles de suggérer une cardiomyopathie sous-jacente. Sur la base d'une suspicion initiale, la mise au point est affinée avec un ECG et, surtout, une imagerie non invasive complète. L'échocardiographie forme la base pour déterminer le phénotype, avec évaluation de l'épaisseur pariétale, de la fonction et des dimensions des ventricules gauche et droit. Le workflow diagnostique réserve par ailleurs une place de premier choix à l'IRM cardiaque (avec injection de gadolinium), qui est conseillée dès l'évaluation initiale de tout patient atteint de cardiomyopathie (classe I). Outre une évaluation minutieuse des volumes, de la fonction et de l'épaisseur pariétale des ventricules gauche et droit, l'IRM a la propriété unique de caractériser les tissus. Et c'est justement ce dernier avantage qui rend l'IRM incontournable dans le diagnostic d'une CNDVG, d'une DVDA, d'une myocardite, d'une amylose, d'une sarcoïdose et d'une hémochromatose. Le profil de fibrose focale, caractérisé par un rehaussement tardif après injection de gadolinium (late gadolinium enhancement, LGE), et/ou de fibrose interstitielle diffuse peut ainsi déjà contenir d'importants indices quant à l'étiologie sous-jacente et par exemple aussi permettre le diagnostic différentiel avec un événement ischémique. Outre l'échocardiographie et l'IRM cardiaque, d'autres modalités d'imagerie peuvent aussi être envisagées, comme la scintigraphie osseuse en cas de suspicion d'amylose ATTR (classe I), le PET-scan en cas de suspicion de sarcoïdose (classe IIa) ou le scanner cardiaque pour exclure une coronaropathie (classe IIa). Une mise au point invasive, par biopsie endomyocardique, n'est conseillée que dans l'éventualité où les résultats d'autres examens suggèrent une myocardite, une infiltration du myocarde ou une cardiomyopathie de surcharge (classe IIa).1
Dès la première présentation sur la mise au point diagnostique, il était évident que le dépistage génétique joue un rôle important dans l'évaluation de chaque patient atteint d'une cardiomyopathie sous-jacente. James Ware (Londres, Royaume-Uni), expert en cardiogénétique, n'a fait que confirmer cette importance lors de la deuxième partie de cette session. La survenue de nombreuses cardiomyopathies dans un contexte familial est universellement connue. En pratique clinique, nous rencontrons le plus souvent le modèle (monogénétique) mendélien, souvent avec transmission autosomique dominante, où une variante rare peut provoquer la maladie. D'autre part, il n'est possible de poser un solide diagnostic monogénétique que dans une minorité des cardiomyopathies (~ 40 % pour la CMH et ~ 30 % pour la CMD). C'est pourquoi il y a de plus en plus de signes indiquant que les cardiomyopathies peuvent aussi avoir une origine polygénétique, à savoir un rapport complexe de différentes variantes plus fréquentes qui, ensemble, peuvent provoquer une pathologie.5
Très tôt dans le processus diagnostique, il est déjà conseillé de s'enquérir des antécédents familiaux et d'établir un arbre généalogique. Un conseil étendu, prodigué par une équipe pluridisciplinaire, suivi d'un dépistage génétique est dès lors vivement recommandé chez tous les patients présentant un diagnostic de novo de cardiomyopathie (classe I). Dès qu'une cause génétique est établie avec certitude (variante de classe 4/5), un dépistage familial en cascade est ensuite conseillé, moyennant des tests cliniques et génétiques, en débutant par les parents au premier degré (classe I). Un dépistage clinique en cascade des membres de la famille est également conseillé en présence d'une variante (vraisemblablement) pathogène, en vue de détecter un éventuel phénotype de cardiomyopathie (classe I). Les tests génétiques permettent par ailleurs aussi de mieux évaluer le risque de répétition dans les générations suivantes et, en cas de désir d'enfant, un dépistage préimplantatoire doit également être envisagé.
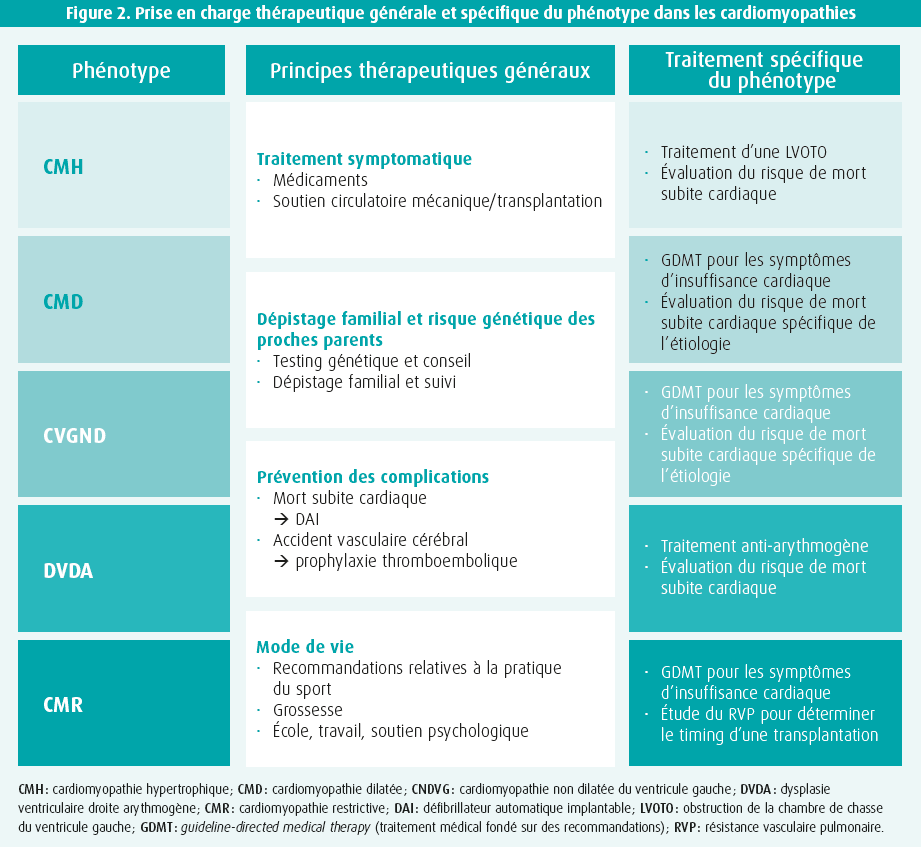
Le génotype sous-jacent, lui aussi, deviendra de plus en plus un facteur déterminant dans la prise en charge thérapeutique. On sait ainsi que différents gènes (FLNC, LMNA, DSP, RBM20…) sont associés à un risque arythmogène plus élevé et, par conséquent, le génotype constitue dans ce cas un argument important en faveur de l'implantation d'un DAI. Pour le reste, ces nouvelles recommandations pour la stratification du risque de mort subite cardiaque chez les patients atteints de CMH reprennent également l'étendue et la présence d'une fibrose myocardique dans la décision d'implanter un DAI. Ainsi, l'implantation d'un DAI peut être envisagée en prévention primaire chez les patients courant un risque de mort subite à 5 ans de < 4 % selon le classique calculateur de risque de MCH6 en présence d'un LGE ≥ 15 % à l'IRM cardiaque (classe IIb).7, 8 Cette même recommandation s'applique aux patients présentant une CMD et une FEVG > 35 %, l'implantation d'un DAI pouvant être envisagée en prévention primaire en présence de facteurs de risque supplémentaires, tels qu'un LGE à l'IRM cardiaque (classe IIb).9, 10 Dans tous ces cas, le choix d'implanter un DAI doit toujours faire l'objet d'une décision partagée entre le médecin et le patient, après discussion de tous les facteurs de risque.1
Pour terminer, Iacopo Olivotto (Florence, Italie) clôture la session en introduisant le concept de prise en charge spécifique du phénotype (figure 2). Les nouvelles recommandations ne se contentent pas de recommandations pour la prévention de la mort subite cardiaque, mais aussi pour un traitement de la fibrillation auriculaire et de l'insuffisance cardiaque adapté au type spécifique de cardiomyopathie (CMH, CMD, CMR…). Dans le cas spécifique de la CMH avec obstacle significatif à l'éjection ventriculaire gauche (gradient ≥ 50 mmHg), le mavacamten - un inhibiteur de l'ATP-ase de la myosine cardiaque - est conseillé lorsqu'un patient reste symptomatique en dépit du traitement par (ou en cas d'intolérance aux) bêtabloquants ou inhibiteurs calciques (classe IIa).11 Enfin, quelques recommandations concernant la pratique sportive dans les différents phénotypes sont encore abordées, l'accent étant mis sur le fait qu'on souhaite autoriser un maximum de patients à avoir une activité physique d'intensité légère à modérée. Chez les patients asymptomatiques présentant une CMH de phénotype bénin, on va encore un peu plus loin et on peut même envisager un exercice physique d'intensité élevée (classe IIb). Dans certains cas spécifiques, comme la DVDA, où le sport peut contribuer à la progression du phénotype, les activités sportives intensives et de compétition restent contre-indiquées (classe III).
En conclusion finale de cette session, l'importance d'un workflow diagnostique multiparamétrique approfondi est encore une fois soulignée, dans l'objectif de pouvoir développer une approche plus personnalisée, fondée sur l'étiologie, pour chacun des patients atteints de cardiomyopathie rencontrés dans notre pratique clinique courante.
Références
- Arbelo, E., Protonotarios, A., Gimeno, J.R., Arbustini, E., Barriales-Villa, R., Basso, C. et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J, 2023.
- Elliott, P.M., Anastasakis, A., Borger, M.A., Borggrefe, M., Cecchi, F., Charron, P. et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J, 2014, 35 (39), 2733-2779.
- Marcus, F.I., McKenna, W.J., Sherrill, D., Basso, C., Bauce, B., Bluemke, D.A. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur Heart J, 2010, 31 (7), 806-814.
- Pinto, Y.M., Elliott, P.M., Arbustini, E., Adler, Y., Anastasakis, A., Böhm, M. et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J, 2016, 37 (23), 1850-1858.
- Tadros, R., Francis, C., Xu, X., Vermeer, A.M.C., Harper, A.R., Huurman, R. et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nature Genetics, 2021, 53 (2), 128-134.
- O'Mahony, C., Jichi, F., Pavlou, M., Monserrat, L., Anastasakis, A., Rapezzi, C. et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur Heart J, 2013, 35 (30), 2010-2020.
- O'Hanlon, R., Grasso, A., Roughton, M., Moon, J.C., Clark, S., Wage, R. et al. Prognostic Significance of Myocardial Fibrosis in Hypertrophic Cardiomyopathy. J Am Coll Cardiol, 2010, 56 (11), 867-874.
- Rowin, E.J., Maron, M.S., Adler, A., Albano, A.J., Varnava, A.M., Spears, D. et al. Importance of newer cardiac magnetic resonance-based risk markers for sudden death prevention in hypertrophic cardiomyopathy: An international multicenter study. Heart Rhythm, 2022, 19 (5), 782-789.
- Mirelis, J.G., Escobar-Lopez, L., Ochoa, J.P., Espinosa, M.Á., Villacorta, E., Navarro, M. et al. Combination of late gadolinium enhancement and genotype improves prediction of prognosis in non-ischaemic dilated cardiomyopathy. Eur J Heart Fail, 2022, 24 (7), 1183-1196.
- Di Marco, A., Brown, P.F., Bradley, J., Nucifora, G., Claver, E., de Frutos, F. et al. Improved Risk Stratification for Ventricular Arrhythmias and Sudden Death in Patients With Nonischemic Dilated Cardiomyopathy. J Am Coll Cardiol, 2021, 77 (23), 2890-2905.
- Olivotto, I., Oreziak, A., Barriales-Villa, R., Abraham, T.P., Masri, A., Garcia-Pavia, P. et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebocontrolled, phase 3 trial. The Lancet, 2020, 396 (10253), 759-769.
Aucun élément du site web ne peut être reproduit, modifié, diffusé, vendu, publié ou utilisé à des fins commerciales sans autorisation écrite préalable de l’éditeur. Il est également interdit de sauvegarder cette information par voie électronique ou de l’utiliser à des fins illégales.