ESC-congresverslag
Naar jaarlijkse traditie werd deze zomervakantie opnieuw afgesloten met het congres van de European Society of Cardiology (ESC), wat dit jaar in Amsterdam plaatsvond van 25 tot 28 augustus 2023. Zoals steeds werden vier nieuwe richtlijnen voorgesteld, waaronder ook de allereerste ESC-richtlijnen voor de aanpak van cardiomyopathieën.1 Naast een grondige update van de (bijna tien jaar oude) richtlijnen over hypertrofe cardiomyopathie2, bevat dit document allemaal volledig nieuwe aanbevelingen over de andere fenotypes. De sessie vond plaats op zondag 27 augustus en werd in goede banen geleid door de twee voorzitters: Elena Arbelo (Barcelona, Spanje) en Juan Pablo Kaski (Londen, Verenigd Koninkrijk).
Iedereen die in de klinische praktijk regelmatig patiënten met cardiomyopathie behandelt, weet dat dit elke keer opnieuw start met een diagnostisch vraagstuk, uitgaande van het fenotype bij presentatie. Er wordt aanbevolen het fenotype te beschrijven aan de hand van enkele morfologische kenmerken zoals linker/rechterventrikelhypertrofie en/ of -dilatatie en daarnaast de eventuele aanwezigheid van een (niet-ischemisch) litteken op MRI. Verder worden ook functionele kenmerken gebruikt bij het definieren van het type cardiomyopathie, zoals systolische en/of diastolische disfunctie van een of beide ventrikels. Naast de gekende hypertrofe (HCM), gedilateerde (DCM) en restrictieve (RCM) cardiomyopathie wordt ook het gebruik van de term 'aritmogene rechterventrikelhypertrofie' (ARVC) opnieuw aangeraden in deze richtlijn.1, 3 Patiënten met een linkszijdige aritmogene cardiomyopathie worden momenteel onder de overkoepelende term 'niet-gedilateerde linkerventrikelcardiomyopathie' (NDLVC) geplaatst. Dat nieuwe fenotype van NDLVC wordt gedefinieerd door de auteurs als 1) de aanwezigheid van niet-ischemische fibrose of infiltratieve ziekte van het linkerventrikel, ongeacht regionale of globale contractiliteitsstoornissen of 2) geïsoleerde linkerventrikelhypokinesie in de afwezigheid van fibrose.4 Daarnaast wordt het fenotype van linkerventrikel non-compactie niet meer beschouwd als cardiomyopathie op zich, maar spreekt men eerder van hypertrabelculatie als een fenomeen dat geïsoleerd kan voorkomen of in combinatie met linkerventrikelhypertrofie, dilatatie en/of systolische disfunctie. Ook het takotsubosyndroom wordt - gezien de voorbijgaande aard van het fenomeen - in de huidige richtlijnen niet meer als cardiomyopathie beschouwd.1
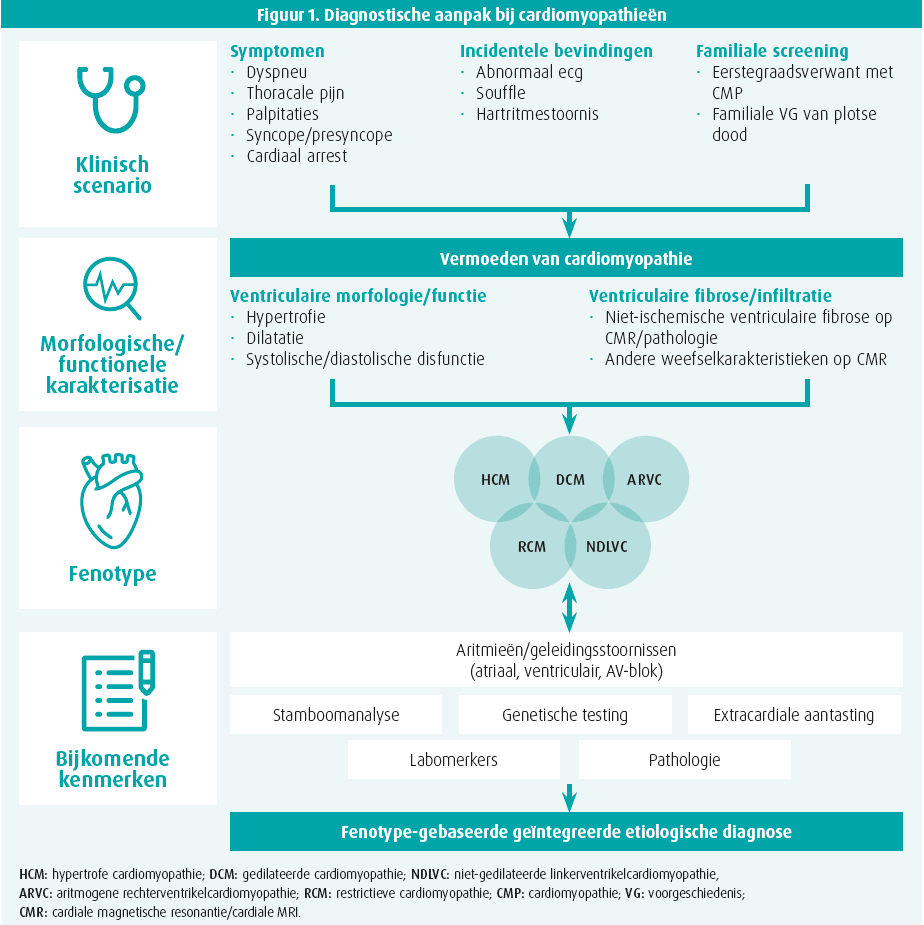
Naast een uitgebreide fenotypische beschrijving van elke cardiomyopathie is een correcte etiologische diagnose cruciaal voor het bepalen van het verdere beleid, al kan dit in de klinische praktijk wel eens een uitdaging vormen. Net daarom wordt er in de richtlijn en ook in deze sessie door Elena Biagini (Bologna, Italië) veel aandacht besteed aan de diagnostische workflow (figuur 1). De eerste stap is zoals steeds een uitgebreide klinische evaluatie, met specifieke aandacht voor symptomatologie, familiale voorgeschiedenis en klinische tekens die suggestief kunnen zijn voor een onderliggende cardiomyopathie. Op basis van een initiële verdenking wordt een verdere uitwerking met een ecg en vooral ook uitgebreide niet-invasieve beeldvorming aangeraden. Echocardiografie vormt de basis voor het bepalen van het fenotype, met evaluatie van de linker- én rechterventrikelwanddikte, -functie en -dimensies. Daarnaast is er in de diagnostische workflow ook een prominente plaats voorzien voor de cardiale MRI-scan (met gadoliniumcontrast), die wordt aangeraden bij de initiële evaluatie van elke patiënt met cardiomyopathie (klasse I). Naast nauwkeurige evaluatie van linker/ rechterventrikelvolumes, -functie en -wanddikte, bevat MRI de unieke eigenschap van weefselkarakterisatie. Net door dat laatste voordeel is MRI een onmisbare factor in de diagnose van NDLVC, ARVC, myocarditis, amyloïdose, sarcoïdose en hemochromatose. Zo kan het patroon van focale fibrose gekenmerkt door late gadolinium enhancement (LGE), en/of diffuse interstitiële fibrose al belangrijke hints bevatten over de onderliggende etiologie en bijvoorbeeld ook de differentiële diagnose met een doorgemaakt ischemisch event mogelijk maken. Naast echocardiografie en cardiale MRI is er ook een plaats voor andere beeldvormingsmodaliteiten, zoals botscintigrafie bij vermoeden van ATTR-amyloïdose (klasse I), PET-CT bij vermoeden van sarcoïdose (klasse IIa) of cardiale CT voor het uitsluiten van coronairlijden (klasse IIa). Invasieve uitweruitwerking door middel van endomyocardiale biopsie wordt enkel aangeraden wanneer de resultaten van andere onderzoeken suggestief zijn voor myocardiale inflammatie, infiltratie of een stapelingsziekte (klasse IIa).1
Al tijdens de eerste presentatie over de diagnostische uitwerking werd duidelijk dat genetische testing een belangrijke rol inneemt bij de evaluatie van elke patiënt met een onderliggende cardiomyopathie. Dat wordt verder verduidelijkt door cardiogenetica-expert James Ware (Londen, Verenigd Koninkrijk) tijdens het tweede deel van deze sessie. Het familiale voorkomen van vele cardiomyopathieën is algemeen bekend. In de klinische praktijk zien we meestal het typisch Mendeliaanse (monogenetische) patroon, vaak met autosomaal dominante overerving, waarbij één zeldzame variant de ziekte kan veroorzaken. Anderzijds kan maar in een minderheid van de cardiomyopathieën een solide monogenetische diagnose worden gesteld (~40 % in HCM en ~30 % in DCM). Daarom zijn er steeds meer aanwijzingen dat cardiomyopathieën ook een polygenetische origine kunnen hebben, zijnde een complexe samenhang van verschillende meer frequente varianten die samen tot pathologie kunnen leiden.5
Al erg vroeg in het diagnostische proces wordt geadviseerd om de familiale voorgeschiedenis na te vragen en een stamboom op te stellen. Uitgebreide counseling door een multidisciplinair team, vervolgd door genetische testing wordt dan ook sterk aangeraden bij elke patiënt met een de novo diagnose van cardiomyopathie (klasse I). Bij zekerheid over een genetische oorzaak (klasse 4/5-variant) wordt vervolgens familiale cascadescreening met klinische en genetische testing aangeraden, startende bij de eerstegraads verwanten (klasse I). Ook in afwezigheid van een (waarschijnlijk) pathogene variant wordt klinische cascadescreening aangeraden bij familieleden om een mogelijk cardiomyopathiefenotype op te sporen (klasse I). Daarnaast kan door genetische testing ook het risico op herhaling in verdere generaties beter worden ingeschat en bij een kinderwens ook pre-implantatie genetische diagnostiek worden overwogen.
Steeds meer zal het onderliggende genotype ook een bepalende factor worden in het therapeutische beleid. Zo is het bekend dat verschillende genen (FLNC, LMNA, DSP, RBM20 …) geassocieerd zijn met een hoger aritmogeen risico en bijgevolg vormt het genotype in dit geval een belangrijk argument voor ICD-implantatie. Verder worden in deze nieuwe richtlijnen voor de risicostratificatie van plotse cardiale dood bij patiënten met HCM ook de uitgebreidheid en aanwezigheid van myocardiale fibrose meegenomen in de beslissing voor ICD-implantatie. Zo kan de implantatie van een ICD in primaire preventie worden overwogen bij patiënten met een vijfjaarsrisico op plotse dood van < 4 % volgens de klassieke HCM-risicocalculator6, indien er op cardiale MRI ≥ 15 % LGE aanwezig is (klasse IIb).7, 8 Dezelfde aanbeveling geldt voor patiënten met DCM en LVEF > 35 % waarbij ICD-implantatie in primaire preventie overwogen kan worden in aanwezigheid van bijkomende risicofactoren zoals LGE op cardiale MRI (klasse IIb).9, 10 In al deze gevallen moet de keuze voor ICD-implantatie steeds een gedeelde beslissing zijn tussen arts en patiënt waarbij alle risicofactoren worden besproken.1
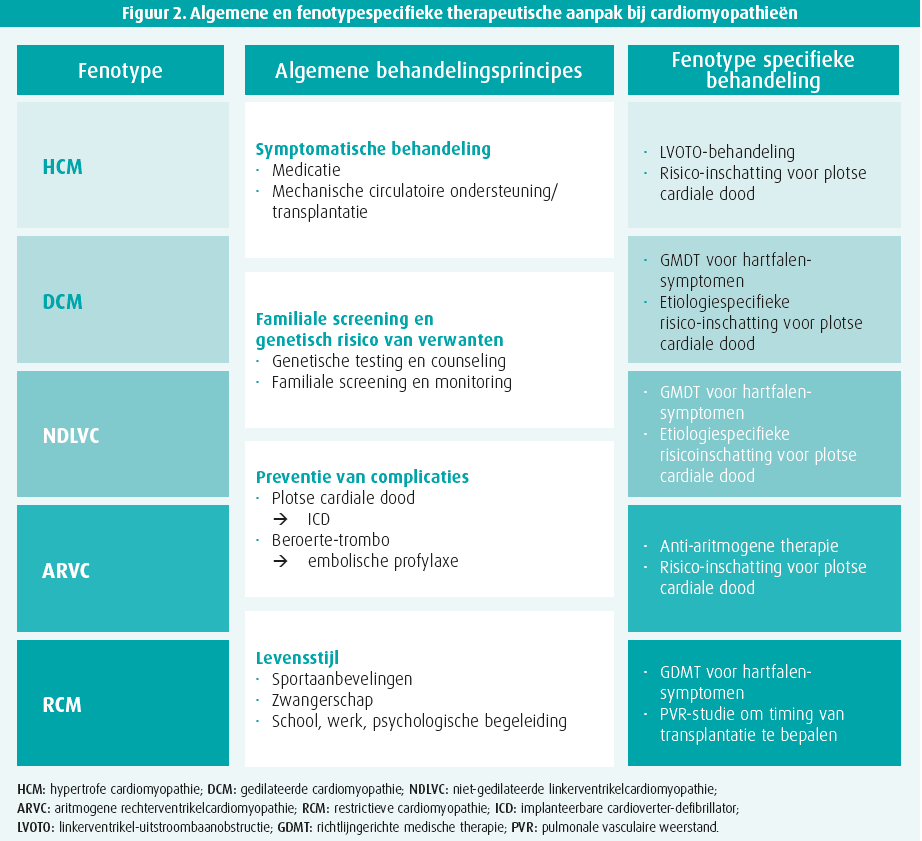
Tot slot wordt de sessie afgesloten door Iacopo Olivotto (Firenze, Italië) die het concept van fenotype-specifiek beleid introduceert (figuur 2). Zo bevatten nieuwe richtlijnen niet enkel aanbevelingen voor preventie van plotse cardiale dood, maar ook voor de behandeling van voorkamerfibrillatie en hartfalen die afgestemd zijn op het specifieke type cardiomyopathie (HCM, DCM, RCM …). Specifiek bij HCM met significante linkerventrikel-uitstroombaanobstructie (gradiënt ≥ 50 mmHg) wordt mavacamten, een inhibitor van cardiale myosine-ATPase, aangeraden als een patiënt symptomatisch blijft ondanks behandeling met (of bij intolerantie voor) bètablokkers of calciumantagonisten (klasse IIa).11 Tot slot worden er nog enkele aanbevelingen besproken rond sportuitoefening bij de verschillende fenotypes met de nadruk op het feit dat men zo veel mogelijk patiënten lichte en matige intensiteit beweging wil toestaan. Bij asymptomatische patiënten met mild HCM fenotype gaat men nog iets verder en kan zelfs beweging aan hoge intensiteit worden overwogen (klasse IIb). In specifieke gevallen zoals ARVC waarbij sport kan bijdragen tot progressie van het fenotype, blijven intensieve en competitieve sportieve activiteiten tegenaangewezen (klasse III).
Als finale conclusie van deze sessie wordt nogmaals het belang van een grondige multiparametrische diagnostische workflow benadrukt, met als doel een meer gepersonaliseerde en op etiologie gebaseerde aanpak te kunnen ontwikkelen voor elke cardiomyopathie patiënt in onze dagelijkse klinische praktijk.
Referenties
- Arbelo, E., Protonotarios, A., Gimeno, J.R., Arbustini, E., Barriales-Villa, R., Basso, C. et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J, 2023.
- Elliott, P.M., Anastasakis, A., Borger, M.A., Borggrefe, M., Cecchi, F., Charron, P. et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J, 2014, 35 (39), 2733-2779.
- Marcus, F.I., McKenna, W.J., Sherrill, D., Basso, C., Bauce, B., Bluemke, D.A. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur Heart J, 2010, 31 (7), 806-814.
- Pinto, Y.M., Elliott, P.M., Arbustini, E., Adler, Y., Anastasakis, A., Böhm, M. et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J, 2016, 37 (23), 1850-1858.
- Tadros, R., Francis, C., Xu, X., Vermeer, A.M.C., Harper, A.R., Huurman, R. et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nature Genetics, 2021, 53 (2), 128-134.
- O'Mahony, C., Jichi, F., Pavlou, M., Monserrat, L., Anastasakis, A., Rapezzi, C. et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur Heart J, 2013, 35 (30), 2010-2020.
- O'Hanlon, R., Grasso, A., Roughton, M., Moon, J.C., Clark, S., Wage, R. et al. Prognostic Significance of Myocardial Fibrosis in Hypertrophic Cardiomyopathy. J Am Coll Cardiol, 2010, 56 (11), 867-874.
- Rowin, E.J., Maron, M.S., Adler, A., Albano, A.J., Varnava, A.M., Spears, D. et al. Importance of newer cardiac magnetic resonance-based risk markers for sudden death prevention in hypertrophic cardiomyopathy: An international multicenter study. Heart Rhythm, 2022, 19 (5), 782-789.
- Mirelis, J.G., Escobar-Lopez, L., Ochoa, J.P., Espinosa, M.Á., Villacorta, E., Navarro, M. et al. Combination of late gadolinium enhancement and genotype improves prediction of prognosis in non-ischaemic dilated cardiomyopathy. Eur J Heart Fail, 2022, 24 (7), 1183-1196.
- Di Marco, A., Brown, P.F., Bradley, J., Nucifora, G., Claver, E., de Frutos, F. et al. Improved Risk Stratification for Ventricular Arrhythmias and Sudden Death in Patients With Nonischemic Dilated Cardiomyopathy. J Am Coll Cardiol, 2021, 77 (23), 2890-2905.
- Olivotto, I., Oreziak, A., Barriales-Villa, R., Abraham, T.P., Masri, A., Garcia-Pavia, P. et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebocontrolled, phase 3 trial. The Lancet, 2020, 396 (10253), 759-769.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.