Deze casus betreft een 81-jarige man met medische voorgeschiedenis van arteriële hypertensie, hypercholesterolemie en percutane coronaire interventie (PCI) van de linker anterior descendens (LAD) na instabiele angineuze klachten.
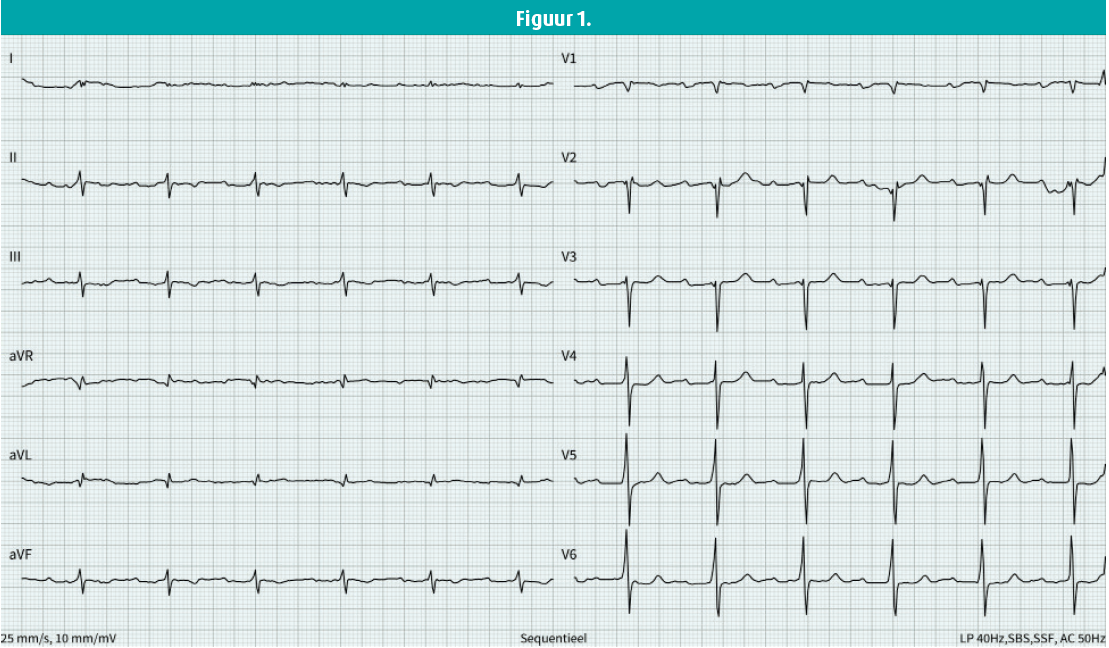
Hij presenteert zich met persisterende vermoeidheidsklachten, orthopneu en progressieve dyspnoeklachten met evolutie tot uiteindelijk NYHA-klasse III. Bij klinisch onderzoek heeft hij een onregelmatig hartritme rond de 70 slagen per minuut en er is geen hartgeruis te horen bij auscultatie. Er zijn discreet tekenen van linkszijdig hartfalen met beperkte aanwezigheid van bilateraal pleuravocht zonder perifere oedemen. Het ecg toont een de novo voorkamerfibrilleren (VKF) met een beeld van een doorgemaakt infarct in het anteroseptaal gebied met QS-beeld in V1 t/m V3. Een controlecoronarografie brengt geen nieuw coronairlijden aan het licht. Er zijn relatief kleine elektrische voltages te zien in de extremiteitsafleidingen (figuur 1). Bij echocardiografie vinden we een concentrische linkerventrikelhypertrofie (LVH) met matig verminderde linkerventrikelfunctie, matig gedilateerde atria en opvallend ook het gespikkelde aspect van het myocard (figuur 2).

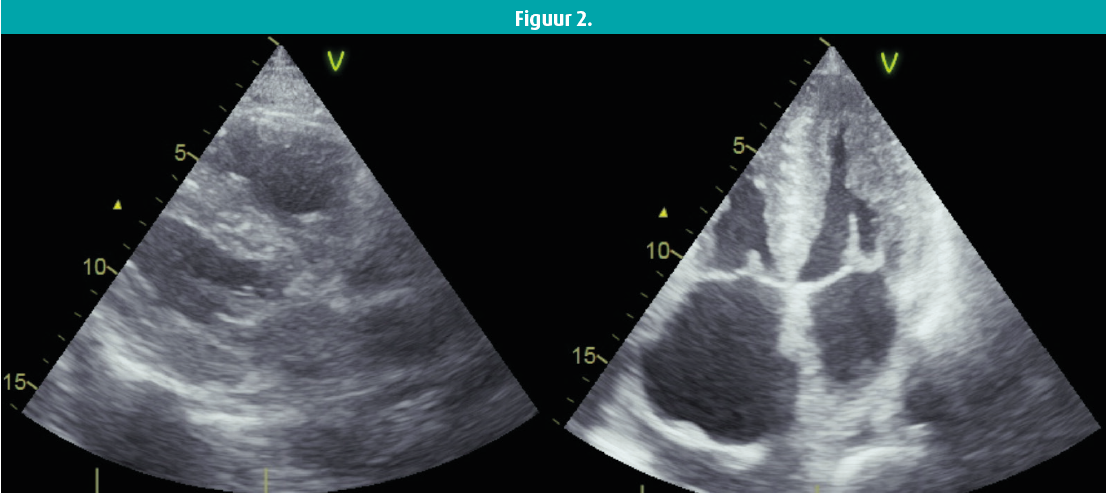
Op basis van bovenstaande bevindingen rijst een sterk vermoeden voor cardiale amyloïdose. De patiënt wordt opgenomen voor verdere diagnostiek en behandeling van het hartfalen. De botscintigrafie is verdacht voor cardiale transthyretineamyloïdose (ATTR). De eiwitelektroforese en serum vrije lichte ketens zijn normaal, wat een cardiale lichteketenamyloïdose uitsluit.
Cardiale ATTR is een infiltratieve aandoening waarbij beschadiging van de myocardcellen optreedt door het neerslaan van amyloïdfibrillen in het hart. Deze fibrillen worden door misgevormde voorloperproteïnen van amyloïd gevormd door een genmutatie. Door infiltratie van deze fibrillen in de myocardcellen ontstaat er een hypertrofe en restrictieve cardiomyopathie. Zowel de incidentie als de prevalentie van cardiale amyloïdose nemen toe. Amyloïdose is een progressieve ziekte en wordt in sommige gevallen ook veroorzaakt door verworven genetische mutaties waardoor de incidentie en prevalentie verder zullen toenemen met de vergrijzende bevolking. Daarnaast wordt de diagnostiek steeds beter door cardiale MRI, botscintigrafie en myocardbiopsie. Nog belangrijker is dat door de klinische aanwijzingen op basis van het klachtenpatroon de afwijkingen op het ecg en TTE sneller kunnen worden herkend.1,2
Gezien de beschadiging aan de myocardcellen en het geleidingssysteem van het hart kunnen veranderingen in het ecg de verdenking op cardiale amyloïdose triggeren. Aanwijzingen op het ecg zijn onder andere aanwezigheid van een eerstegraads AV-blok (31 %), bundeltakblok (15-17 %), voorkamerfibrillatie/-flutter (56 %), microvoltages (22 %, ≤ 1,0 mV in de precordiale en ≤ 0,5 mV in de extremiteitenafleidingen), LVH (11 %), disconcordantie tussen de grootte van de voltages op het ecg met de werkelijke ventrikelhypertrofie en pseudo-infarctpatroon (63 %, voornamelijk in het anterior gebied).3
Zo zien we op het ecg van onze patiënt van zes maanden voordien, met toen nog milde klachten, een sinusritme met uitgesproken eerstegraads AV-blok (PR-interval van 362 ms) en microvoltages in de extremiteitenafleidingen (figuur 3).
De behandeling van cardiale ATTR is enerzijds gericht op de behandeling van hartfalen en de onderliggende aritmie. Anderzijds bestaat er naast de klassieke hartfalentherapie ook een specifieke medicamenteuze therapie die het ziekteproces van cardiale ATTR kan afremmen. Tafamidis is een transthyretinestabilisator die de vorming van amyloïdfibrillen kan tegengaan en die sinds oktober 2021 in België wordt terugbetaald. In het algemeen is de prognose van cardiale ATTR slecht, met een gemiddelde overleving van twee tot drie jaar zonder adequate behandeling.4-5 Voor de prognose is het belangrijk in welk stadium de ziekte wordt vastgesteld en wanneer adequate therapie wordt opgestart. Daarom is het vroegtijdig herkennen van cardiale amyloïdose, met onder andere herkenning van de aanwijzingen op het ecg belangrijk om zo snel mogelijk therapie te starten.
Referenties
- Patel, K.S., Hawkins, P.N. Cardiac amyloidosis: where are we today? J Intern Med, 2015, 278 (2), 126-144.
- Gilstrap, L.G., Dominici, F. Wang, Y., El-Sady, M.S., Singh, A., Di Carli, M.F. et al. Epidemiology of Cardiac Amyloidosis Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ Heart Failure, 2019, 12 (6), e005407.
- González-López, E., Gagliardi, C., Dominguez, F., Quarta, C.C., Haro-Del Moral, F.J., Milandri, A. et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J, 2017, 38 (24), 1895-1904.
- Maurer, M.S., Schwartz, J.H., Gundapaneni, B., Elliot, P.M., Merlini, G., Waddington-Cruz, M. et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med, 2018, 379 (11), 1007-1016.
- Lane, T., Fontana, M., Martinez-Naharro, A., Quarta, C.C., Whelan, C.J. Petrie, A. et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation, 2019, 140 (1), 16-26.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.