Compte rendu d'une session de la BSC - session 1 - BWGACHD
Cet article rend compte d'une session organisée par le BWGACHD (Groupe de travail belge pour les adultes atteints d'une cardiopathie congénitale) lors du congrès 2021 en ligne de la BSC.
Ces dix dernières années ont apporté de nouvelles connaissances et des avancées dans les soins, plus spécifiquement en matière d'interventions percutanées, d'évaluation des risques et de timing d'une intervention ou d'un traitement chirurgical ainsi que de politique médicamenteuse. Le moment de nouvelles directives était donc venu. Les directives 2020 de l'ESC pour les adultes atteints d'une cardiopathie congénitale (ACHD) insistent sur le processus décisionnel, composé d'un diagnostic correct, du timing de l'intervention (percutanée), de l'évaluation des risques et du choix de l'intervention la plus adaptée. Des conditions spécifiques, telles que l'insuffisance cardiaque, l'hypertension pulmonaire et l'utilisation d'anticoagulants, sont également abordées. Lors de cette session de la BSC, nous avons la chance que trois orateurs belges, tous experts internationaux dans le domaine de l'ACHD, viennent expliquer divers aspects des nouvelles directives. C'est avec une grande fierté que le BWGACHD présente Julie De Backer, qui s'est énormément investie en tant que coprésidente ces dernières années afin de donner corps à ces nouvelles directives.
Directives ACHD de l'ESC : qu'est-ce qui a changé en une décennie ?
Julie De Backer, UZ Gent
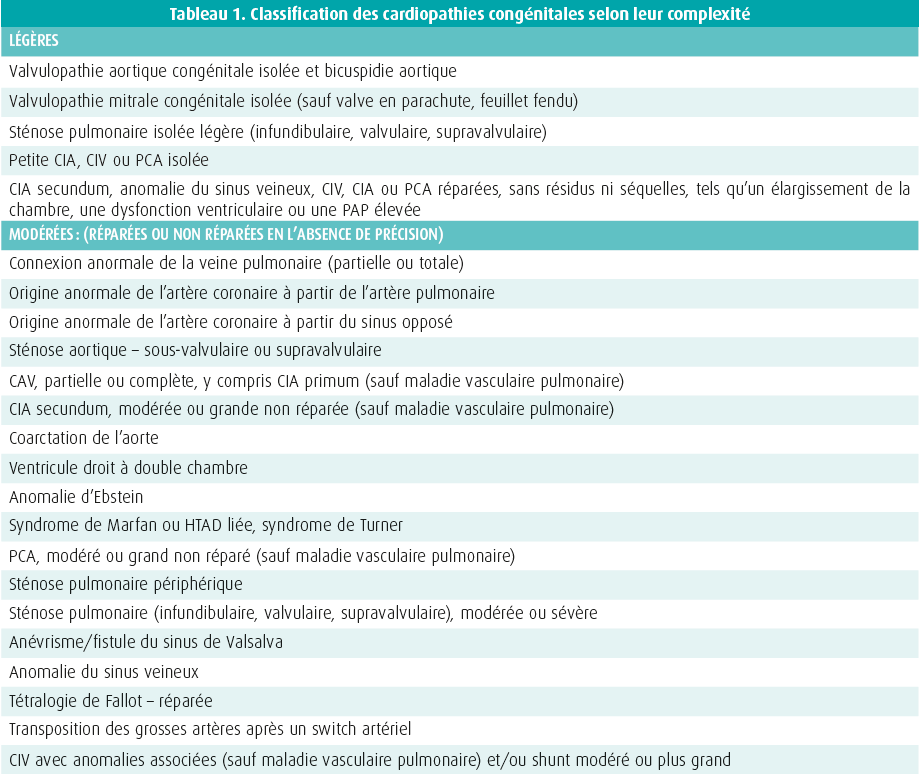
Depuis 2010, beaucoup de choses ont changé en ce qui concerne les soins des patients atteints d'une cardiopathie congénitale. Le changement le plus marquant est probablement la modification de la dénomination grown-up with congenital heart disease (GUCH) en adults with congenital heart disease (ACHD). Le terme « grown-up » est en effet utilisé pour les adolescents, alors que de plus en plus de patients ont désormais atteint l'âge adulte. Comme le montre la figure 1, le message central est le suivant : une cardiopathie congénitale est considérée comme une affection chronique, qui mérite l'attention et les soins nécessaires tout au long de la vie du patient. Le deuxième message clé est que l'ACHD ne doit pas être considérée comme une entité unique, mais comme un ensemble complexe de troubles. La complexité dépend ici de l'anomalie anatomique initiale. Les patients ACHD sont dès lors classés en trois catégories : cardiopathies légères, modérées et complexes (tableau 1). Les directives se composent d'abord de conseils généraux, puis décrivent en détail quelques-unes des anomalies les plus fréquentes.
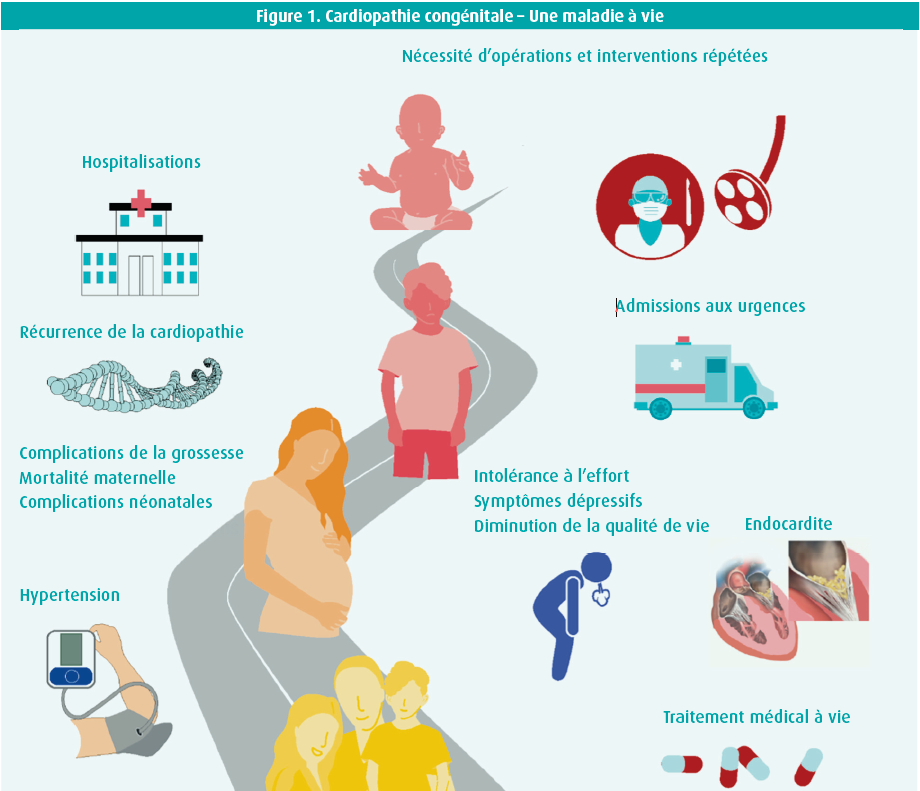
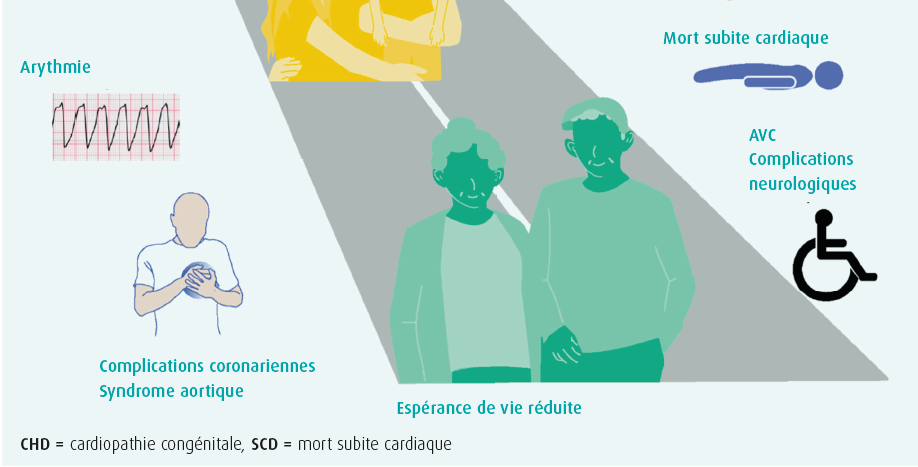

Des recommandations spécifiques à l'intention des professionnels de la santé sont décrites en ce qui concerne l'organisation des soins des patients ACHD et se distinguent par leur caractère multidisciplinaire. La transition de la cardiologie pédiatrique vers la cardiologie pour adultes exige une attention et une organisation spécifiques.
Quelques considérations thérapeutiques
Un paragraphe relatif aux troubles du rythme a été ajouté, car ils sont une cause de mortalité importante dans l'ACHD. Le principal message est ici le suivant : comprendre la cause, le mécanisme et l'anatomie de la cardiopathie congénitale sous-jacente (CHD). Une approche multidisciplinaire est essentielle pour un traitement optimal des troubles du rythme. En cas de tachycardie supraventriculaire symptomatique, une ablation précoce par cathéter doit être envisagée, comme alternative à un traitement médicamenteux à vie. Chez les patients atteints d'une tétralogie de Fallot avec tachycardie ventriculaire (TV) dans la région de la chambre de chasse du ventricule droit (CCVD), il est recommandé de procéder à une ablation de la TV avant un remplacement de valve, car le remplacement valvulaire aussi bien percutané qu'aortique rend les substrats de la TV inaccessibles par la suite.
En ce qui concerne le traitement par anticoagulants, les recommandations suivantes s'appliquent : chez les patients atteints d'une CHD légère, les scores CHA2DS2-VASC et HAS-BLED sont utilisés comme parmi la population générale. L'anticoagulation est recommandée en cas de fibrillation atriale paroxystique et persistante dans la CHD modérée et sévère, mais une approche individualisée reste nécessaire. L'utilisation d'anticoagulants oraux (OAC) en cas de circulation de Fontan reste peu claire. Les nouveaux OAC (NOAC) semblent sûrs en tant qu'alternative aux antagonistes de la vitamine K, en l'absence de valve artificielle ou de sténose mitrale grave. Un registre récent en Allemagne a toutefois mis en évidence des résultats plutôt négatifs pour les NOAC. Davantage de données sont donc nécessaires.
Recommandations supplémentaires
Advanced care planning
Ce sujet bénéficie de toujours plus d'attention en raison de l'amélioration de la survie des patients ACHD atteints de troubles complexes. Les recommandations ont été amplement décrites dans un document distinct3 et soulignent l'importance d'élaborer en temps opportun un plan de soins dans lequel le patient occupe une position centrale.
Pregnancy, anticonception and genetic counseling
Les jeunes présentant une ACHD atteignent de plus en plus souvent l'âge auquel ils pensent à avoir eux-mêmes des enfants. La grossesse et les malformations cardiaques ont été décrites en 2018 dans des directives distinctes. Les directives actuelles contiennent toutefois un tableau qui indique les circonstances dans lesquelles une grossesse représente un risque élevé et les cas où le risque est tellement élevé que la grossesse est déconseillée. La nouveauté de ces directives est qu'elles indiquent le risque de transmission de la maladie, par affection et par sexe.
Affections spécifiques
Les modifications importantes par rapport aux directives précédentes sont expliquées brièvement par affection. Quelques messages à retenir sont résumés ci-dessous :
- Shunt : une mesure invasive de la résistance vasculaire pulmonaire est nécessaire chez les patients présentant un shunt et des signes de pression pulmonaire accrue.
- La fermeture percutanée d'une CIV (résiduelle) est une alternative possible à la chirurgie.
- (Re)coarctation aortique : une mesure invasive de la pression est nécessaire. Un traitement percutané avec pose de stent est privilégié. Une autre nouveauté est que l'intervention doit être envisagée également chez les patients normotendus lorsque le gradient de pression augmente.
- Aortopathie : le groupe de l'aortopathie a été étendu du syndrome de Marfan aux aortopathies familiales. La bicuspidie et le syndrome de Turner ont été ajoutés, ainsi que les facteurs de risque génétiques spécifiques connus (TGFBR1/2). Un remplacement du bulbe aortique, qui épargne la valve, par un chirurgien expérimenté, est recommandé parmi cette population jeune.
- Tétralogie de Fallot : le seuil de l'intervention varie en fonction de la possibilité de fermeture percutanée ou chirurgicale. Des critères de volume du ventricule droit sont également pris en considération. Le remplacement valvulaire percutané est privilégié dans la CCVD non native. Le traitement de l'arythmie éventuelle doit faire l'objet d'une concertation multidisciplinaire préalable.
- Circulation de Fontan : les troubles persistants du rythme atrial sont une urgence médicale et doivent se traiter sans attendre au moyen d'une cardioversion électrique. Des anticoagulants sont recommandés en cas de thrombus atrial, de troubles du rythme ou d'événements thromboemboliques. Même lorsqu'une seule complication est présente, la grossesse est déconseillée. Enfin, le seuil du recours à des mesures invasives de la pression en cas de dégradation clinique est bas.
Message clé des directives 2020 de l'ESC :
Tous les patients ACHD doivent être vus au moins une fois par un expert de l'ACHD, afin de déterminer le plan de suivi optimal. L'insuffisance cardiaque, les troubles du rythme, l'hypertension pulmonaire, la cyanose et la grossesse exigent l'avis d'un expert. L'ACHD forme un groupe très varié en termes de complexité. Chaque patient est différent. Une approche individualisée, souvent multidisciplinaire, est nécessaire.
Recueillir des preuves au-delà des directives : mises en garde et opportunités
Alexander Van De Bruaene, UZ Leuven
L'objectif de cette présentation est de donner un aperçu, à l'aide de quelques exemples, de la force des preuves scientifiques dans le domaine des cardiopathies congénitales et de l'importance des registres cliniques. Le groupe des ACHD rassemble en effet une population très hétérogène. En outre, lors d'une étude, il est souvent difficile de déterminer des critères de jugement concrets ou de substitution pour des paramètres cliniquement importants, des modèles animaux spécifiques ne sont pas disponibles et les études de phase 3 parmi de grands groupes de patients sont pour ainsi dire irréalisables. Les nouvelles directives en témoignent : une seule recommandation a reçu le niveau A et seulement trois ont reçu le niveau B, toutes les autres directives reposent sur des opinions d'experts, de petites études rétrospectives et des registres (niveau C).
La seule étude randomisée contrôlée (sur laquelle repose le seul niveau de preuve A), initialement conçue comme étude de sécurité, a évalué l'effet du bosentan contre placebo, chez 54 patients atteints du syndrome d'Eisenmenger randomisés selon un rapport de 2:1. Un effet positif du traitement et une résistance vasculaire pulmonaire ont été mises en évidence au test de marche de 6 minutes. Quelques années plus tard, un effet positif du traitement sur la mortalité a été démontré sur la base d'un registre clinique à l'hôpital Royal Brompton. Les bases de données cliniques peuvent fournir des informations sur le suivi des patients au fil des ans. L'avantage est ici que des associations peuvent souvent être déterminées entre les caractéristiques de base et les paramètres des résultats et que les données relatives au traitement des patients et aux facteurs de risque sont complètes. L'inconvénient est toutefois que, contrairement à une étude randomisée contrôlée, les caractéristiques et les profils de risque diffèrent entre les groupes de traitement et qu'une analyse statistique spécifique, tenant compte également des facteurs exerçant éventuellement une influence, est nécessaire.
L'Euro Heart Survey, dans laquelle, au total, 4 110 patients de 26 pays ont été enregistrés, et qui évalue surtout le traitement médical et l'utilisation des infrastructures de santé est un exemple important de base de données cliniques. Barbara Mulder a ensuite mis sur pied un projet similaire (le registre CONCOR), aux Pays-Bas, afin d'examiner de plus près la mortalité. Ce projet est un grand succès, avec, entre-temps, 17 000 patients et 70 publications. Cet exemple ne fait que confirmer à quel point les bases de données cliniques peuvent être précieuses. La Brompton Database on Congenital Heart Disease est un autre exemple. Depuis trente ans, elle rassemble les données de plus de 70 000 patients d'un seul centre. L'analyse de la mortalité révèle que l'insuffisance cardiaque est la cause principale de décès dans l'ACHD.
Les bases de données cliniques peuvent également s'utiliser pour répondre à une question spécifique. Une des questions les plus brûlantes est le moment correct du remplacement de la valve pulmonaire chez les patients asymptomatiques présentant une tétralogie de Fallot. Les méta-analyses de plusieurs bases de données cliniques présentent ici des informations afin d'étayer les directives cliniques. Un deuxième domaine dans lequel les bases de données cliniques ont fourni de nombreuses informations est celui des registres des grossesses (initialement ZAHARA et CARPREG, actuellement ROPAC, un registre européen sous la direction de Jolien Roos-Hesselink). Les bases de données peuvent parfois montrer des informations contradictoires, par exemple si l'usage des NOAC est sûr, il possède néanmoins plus d'effets indésirables dans l'ACHD. Enfin, les bases de données cliniques peuvent aussi s'employer comme indicateur de la qualité. Ainsi, l'utilisation d'un registre peut être à l'origine de davantage de renvois vers un centre ACHD et avoir un impact favorable à long terme. En résumé, nous pouvons conclure que la force des preuves dans l'ACHD est tout au plus modérée, principalement en raison d'une population de patients hétérogène dont les sous-groupes ne comportent qu'un petit nombre de personnes. Les registres prospectifs fournissent des informations précieuses pour la pratique clinique et les résultats des paramètres.
Soins de l'ACHD : un processus à vie
Agnès Pasquet - UCL Saint-Luc Bruxelles
L'amélioration de la survie au cours des dernières décennies fait que nous rencontrons aujourd'hui de plus en plus de patients adultes présentant une cardiopathie congénitale (ACHD). En Europe, 2012 a été un tournant important en la matière. En effet, c'est à partir de ce moment que les patients adultes présentant une cardiopathie congénitale sont devenus plus nombreux que les enfants présentant une cardiopathie congénitale. Ce groupe de patients ACHD ne va faire qu'augmenter. Mieux encore, il existe même un groupe sans cesse plus grand de patients ACHD âgés de plus de 60 ans. Les soins de ce groupe de patients sont un processus à vie, au cours duquel chaque phase ou chaque facteur supplémentaire peut avoir un impact important sur l'espérance de vie (figure 1). Les causes de décès sont principalement déterminées par l'affection cardiovasculaire, l'insuffisance cardiaque et la mort subite associée aux troubles du rythme cardiaque ainsi que le décès post-opératoire précoce. Quelquesuns des facteurs ayant le plus d'influence sont expliqués plus amplement dans cette présentation. À commencer par l'insuffisance cardiaque. L'incidence varie de 20 à 50 %, selon la cardiopathie sous-jacente. L'observation selon laquelle une hospitalisation liée à une insuffisance cardiaque augmente de 35 % le risque de décès au cours des quatre années suivant cette hospitalisation est toutefois encore plus importante. La pathophysiologie sous-jacente est ici multifactorielle et est notamment déterminée par l'anatomie, les malformations résiduelles, les problèmes acquis supplémentaires tels qu'une ischémie et des troubles du rythme, etc. Outre les symptômes classiques de l'insuffisance cardiaque, celle-ci peut s'exprimer par des troubles du rythme (ventricule droit systémique, par exemple), une mort subite (dans la tétralogie de Fallot, par exemple) et une diarrhée (circulation de Fontan associée à une perte de protéines gastro-intestinale) dans l'ACHD. Poser un diagnostic correct peut donc se révéler un défi. Le pronostic est également influencé de manière négative par un âge avancé, une maladie rénale, une insuffisance hépatique et une ischémie cardiaque ou cérébrale. Le point important suivant est de prévenir les troubles du rythme. Selon la base de données CONCOR, ils surviennent chez 25 % de tous les patients ACHD aux Pays- Bas. Le pourcentage de patients présentant des troubles du rythme augmente avec l'âge, mais le plus préoccupant est la mort subite liée à une arythmie ventriculaire dans 7 à 26 % de tous les décès de patients adultes, en fonction de l'affection sous-jacente. Le risque d'endocardite parmi la population ACHD est plus élevé que parmi la population générale, notamment chez les patients après une chirurgie valvulaire, une endocardite antérieure et certains conduits. Une attention spécifique est nécessaire pour les patients porteurs d'une prothèse valvulaire en position pulmonaire, comme après l'implantation d'une valve Melody, lors de laquelle une incidence atteignant 5,7 % par personne-année a été décrite. La nécessité d'une ré-opération (notamment dans la tétralogie de Fallot, la coarctation aortique, la transposition des gros vaisseaux et Fontan) influence chaque fois l'espérance de vie. Les troubles neurovasculaires sont également un point à surveiller à vie. Depuis le foetus et le nourrisson, avec l'exposition à l'hypoxie, puis les modifications hémodynamiques et la cyanose ou l'influence de la machine coeur-poumon sur un enfant, jusqu'à l'apparition de facteurs de risque supplémentaires tels que la fibrillation atriale en tant qu'ACHD… Tous ces facteurs peuvent conduire à des perturbations du développement neurologique, à des lésions cérébrales et à un retard neurocognitif. À cet égard, il est également à noter que la démence est plus fréquente parmi la population ACHD. Le risque d'infarctus cérébral est plus élevé et augmente en cas de shunt résiduel ainsi qu'en présence d'une insuffisance cardiaque. En plus de tous ces facteurs de risque cardiovasculaires, la charge des comorbidités cardiovasculaires et non cardiovasculaires, qui influencent également la survie, est importante.
Le message clé de cette présentation est que les patients ACHD doivent être suivis comme tout autre patient chronique. Les complications possibles, notamment l'insuffisance cardiaque, les troubles du rythme et les maladies cérébrovasculaires, doivent être dépistées avec soin. En outre, les facteurs de risque classiques et les comorbidités qui accroissent le risque cardiovasculaire parmi cette population augmentent avec l'âge.
Références
- Baumgartner, H., De Backer, J. et al 2020 ESC Guidelines for the management of adult congenital heart disease: The Task Force for the management of adult congenital heart disease of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Adult Congenital Heart Disease (ISACHD), Eur Heart J, 2021, 42 (6), 563-645.
- Baumgartner, H., De Backer, J. The ESC Clinical Practice Guidelines for the Management of Adult Congenital Heart Disease 2020. Eur Heart J, 2020, 43, 4153- 4154.
- Schwerzmann, M. et al. Recommendations for advance care planning in adults with congenital heart disease: a position paper from the ESC Working Group of Adult Congenital Heart Disease, the Association of Cardiovascular Nursing and Allied Professions (ACNAP), the European Association for Palliative Care (EAPC), and the International Society for Adult Congenital Heart Disease (ISACHD). Eur Heart J, 2020, 41, 4200-4210.
- Engelfriet, P. et al The spectrum of adult congenital heart disease in Europe: morbidity and mortality in a 5 year followup period. The Euro Heart Survey on adult congenital heart disease. Eur Heart J, 2005, 26 (21), 2325-2333.
- Baumgartner, H. Geriatric congenital heart disease: a new challenge in the care of adults with congenital heart disease? Eur Heart J, 2014, 35 (11), 683-685.
- Raissadati, A. et al. Late Causes of Death After Pediatric Cardiac Surgery: A 60-Year Population-Based Study. J Am Coll Cardiol, 2016, 68 (5), 487-498.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.