Naar aanleiding van het satellietsymposium 'Genetic diseases in cardiology: approach in your daily practice' is het onderwerp zeldzame hartziekten aangesneden op het 37e congres van de BSC. We mochten twee experts ter zake verwelkomen, namelijk professor P. Elliott en professor A. Linhart, die de discussie gevoerd hebben samen met professor Julie de Backer en professor Antoine Bondue, de moderatoren van de sessie.
The cardiologist facing rare diseases in clinical practice: be aware!
Professor P. Elliott is van het University College London in het Verenigd Koninkrijk naar België gekomen om ons duidelijk te maken dat het belangrijk is te denken aan een 'zeldzame' hartziekte bij een patiënt met 'atypische' verschijnselen. Hij benadrukte dat elke cardioloog in zijn praktijk patiënten met een zeldzame ziekte kan tegenkomen. Hij moet die dus kennen en erop bedacht zijn.
Professor Elliott belichtte eerst een belangrijke paradox inzake zeldzame ziekten: een ziekte wordt als zeldzaam beschouwd als de prevalentie minder dan 1 op de 2000 bedraagt. Het aantal ziektes dat in die categorie valt, is echter vrij hoog, in die mate zelfs dat tot 8 % van de bevolking een 'zeldzame' ziekte zou kunnen hebben. In Europa kennen we 5000 tot 8000 'zeldzame' ziektes, die voorkomen bij in totaal 36 miljoen mensen. De meeste (80 %) zijn van genetische oorsprong, zijn chronisch en kunnen fataal aflopen. Het zijn dus potentieel ernstige ziektes. Driekwart van de patiënten vertoont symptomen vanaf de kindertijd. Als iemand op jonge leeftijd symptomen ontwikkelt, moet je dus zeker denken aan een zeldzame ziekte. Een klinisch kenmerk van 'zeldzame' ziekten is dat de diagnose laat wordt gesteld, hoofdzakelijk bij gebrek aan kennis, omdat we er niet op letten, omdat we andere leden van de familie niet onderzoeken en omdat de patiënt vaak laat wordt verwezen naar een referentiecentrum.
Statistische gegevens die in 2012 in de JACC1 gepubliceerd zijn, leren dat meer dan de helft van de sterfte aan verworven ziekten in de ontwikkelde landen toe te schrijven is aan hart- en vaataandoeningen. Die laatste correleren sterk met goed bekende risicofactoren. Door aanpak van die risicofactoren en doeltreffende preventiecampagnes is de incidentie van hart- en vaataandoeningen de laatste 30 jaar duidelijk gedaald.
Is het dan gewettigd te zoeken naar zeldzame en erfelijke ziekten? Professor Elliott heeft ons daar subtiel van overtuigd aan de hand van enkele klinische voorbeelden. De uitdaging ligt er volgens hem in om tussen al de patiënten die in de dagelijkse praktijk gezien worden, de patiënten met een 'zeldzame' en mogelijk hereditaire hartziekte te identificeren. Bij een 'zeldzame' ziekte is er soms een familiaire voorgeschiedenis, maar een 'sporadische' presentatie sluit een erfelijke aandoening niet uit. We kennen bijvoorbeeld nu al meer dan 50 genen die geassocieerd zijn met een gedilateerde cardiomyopathie, met meestal een mendeliaanse overerving. Als je een systematische klinische screening uitvoert bij de naaste familieleden met een 'sporadische' gedilateerde cardiomyopathie, vind je in 15-25 % van de gevallen klinische of subklinische afwijkingen (naargelang van het gebruikte diagnostische criterium en de onderzochte populatie), wat er dus op wijst dat het in feite gaat om een 'hereditaire' aandoening. Volgens een recent Fins register2 kunnen we op dit ogenblik een mutatie terugvinden bij 48 % van de patiënten met een gedilateerde cardiomyopathie met een familiaire voorgeschiedenis en bij 24 % van de patiënten met een gedilateerde cardiomyopathie zonder familiaire voorgeschiedenis. Die gegevens worden op grotere schaal bevestigd door het Europese register3. Dat wijst op een belangrijke genetische component bij het optreden van een gedilateerde cardiomyopathie, met evenwel een onvolledige penetrantie (dat betekent dat nog andere factoren meespelen, zoals de omgeving, de levenswijze, epigenetische veranderingen en modulerende genen).
In de kliniek is het belangrijk om alarmverschijnselen, 'red flags' genoemd, te herkennen, die ons op het spoor kunnen zetten van een erfelijke aandoening of bepaalde mutaties. De richtlijnen voor de behandeling van hartfalen zijn van een uitstekende kwaliteit, maar bij het zoeken naar de oorzaak van het hartfalen beperken ze zich tot de klassieke aandoeningen en hebben ze het minder vaak over erfelijke ziekten. De arts moet dus een grondige anamnese uitvoeren en goed letten op tekenen die zouden kunnen wijzen op een genetische aandoening (jonge leeftijd, familiaire antecedenten, echocardiografische kenmerken, comorbiditeit, extracardiale verschijnselen …). Op grond daarvan kan hij de patiënten selecteren bij wie een aanvullende evaluatie gewettigd is. Sommige mutaties resulteren in een speciale klinische evolutie. Mutaties van Lamin A/C4 of FLNC5 bijvoorbeeld veroorzaken vaak ritmestoornissen. Die patiënten moeten dan ook agressiever behandeld worden. Vandaar ook het concept van 'gestratificeerde geneeskunde' of 'geneeskunde op maat'. Dat impliceert het identificeren van subgroepen van patiënten die baat zouden kunnen vinden bij een specifiekere behandeling.
Om een diagnose te kunnen stellen van die 'zeldzame' ziekten (die echter alles samen genomen toch frequent zijn), moeten we er allen goed op bedacht zijn. Met een familiaire anamnese, herkenning van suggestieve tekenen en een aandachtig klinisch onderzoek (om altijd een verklaring te zoeken voor 'atypische' symptomen) kun je een 'zeldzame' ziekte herkennen in de dagelijkse praktijk. De genetica van die aandoeningen is vaak complex. Een nauwe samenwerking met centra voor menselijke erfelijkheid is overigens vereist om de juiste diagnose te kunnen stellen en om die patiënten en hun familie optimaal te behandelen.
Emerging therapies for hereditary cardiomyopathies
De tweede sessie van het symposium werd gepresenteerd door prof. A. Linhart. van het Universitaire Ziekenhuis van Praag. Net als zijn voorganger legde hij de eerste minuten van zijn uiteenzetting uit hoe belangrijk het is om te zoeken naar tekenen die wijzen op een zeldzame ziekte, en daarna ging hij in op de nieuwe therapeutische mogelijkheden voor die patiënten.
Elke cardiologische evaluatie omvat basisonderzoeken (ecg en echocardiografie) en een zo volledig mogelijke persoonlijke én familiaire anamnese. Aan de hand van de familiaire anamnese en een grondig klinisch onderzoek kun je een erfelijk patroon herkennen en/of een aantasting van het hart of andere organen vaststellen bij de patiënt en diens naaste verwanten. Diverse aanvullende onderzoeken zullen dan uitmonden in de finale diagnose.
Diagnostisch beleid
Een grondige analyse van het elektrocardiogram toont vaak bepaalde oorzaken in geval van speciale kenmerken. Tabel 1 geeft daar een voorbeeld van voor ventrikelhypertrofie.
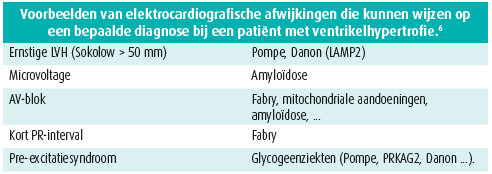
Het is belangrijk een stamboom op te stellen om de wijze van overerving te bepalen. De stamboom kan ons op het spoor van bepaalde aandoeningen zetten. Zo zijn de ziekte van Fabry en de ziekte van Danon X-gebonden aandoeningen. Mitochondriale aandoeningen worden hoofdzakelijk overgedragen door de moeder.
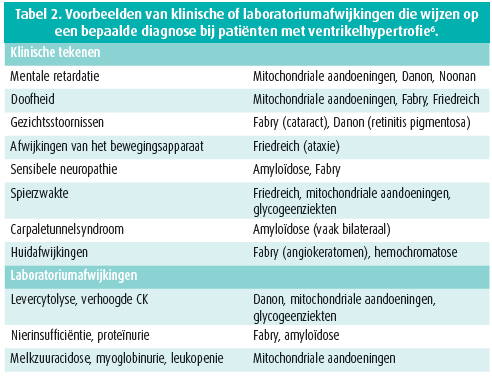
Je moet ook bijzonder goed letten op afwijkingen elders. Die zijn soms zeer invaliderend, maar vaak discreet en geleidelijk opkomend. Om het even welk orgaan kan worden aangetast, met name de huid, het zenuwstelsel en de ogen, en er kunnen laboratoriumafwijkingen optreden. Dat kan ons op het spoor van bepaalde diagnosen zetten en het is een essentieel element in het diagnostische beleid. Tabel 2 is een onvolledige lijst van de klinische tekenen en laboratoriumafwijkingen die kunnen optreden bij patiënten met een genetisch bepaalde ventrikelhypertrofie.
Een echocardiografie is de hoeksteen bij het diagnosticeren van cardiomyopathie, maar een MRI van het hart is ook bijzonder interessant. Het is een niet-invasief onderzoek dat informatie geeft over de weefselkenmerken van het myocard en functionele elementen. Met een MRI van het hart kun je de ventrikelvolumes berekenen en de ventrikelfunctie goed evalueren. Na injectie van gadolinium kun je weefselveranderingen evalueren en met name zones van fibrose lokaliseren (late aankleuring, Late Gadolinium Enhancement of LGE, als gevolg van een significante toename van de lavagetijd van de fibrotische cellen in vergelijking met gezond weefsel). De fibrose die optreedt na een myocardinfarct, volgt de anatomie van de kransslagaders. Bij een niet-ischemische cardiomyopathie ziet de aankleuring er anders uit7, 8.
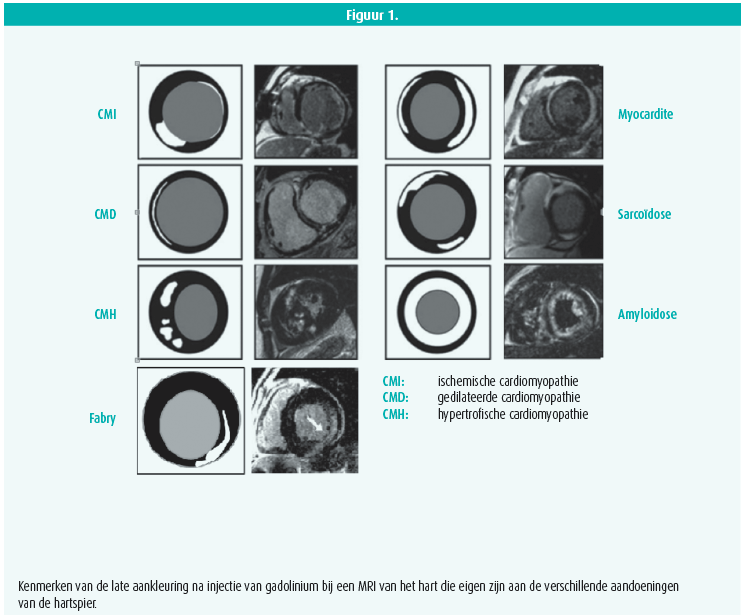
Prof. Linhart had het ook over het belang van de T1-mappingtechniek. Met die techniek kun je de differentiële diagnose van cardiomyopathie stellen via analyse van de intrinsieke weefselkenmerken van het myocard of na injectie van gadolinium. Met die veelbelovende techniek kun je het extracellulaire volume berekenen en patronen herkennen die specifiek zijn voor de verschillende oorzaken van cardiomyopathie. Je zou het haast kunnen vergelijken met een niet-invasief 'histologisch' onderzoek van het myocardweefsel. Dankzij die innovatieve technieken kunnen we afzien van een myocardbiopsie9, 10.
Gerichte behandelingen
Met een betere kennis van de moleculaire mechanismen kan de identificatie van een mutatie in een metabole weg of die invloed heeft op een fysiologisch proces, ons op het spoor zetten van 'gerichte' geneesmiddelen, die de natuurlijke evolutie van bepaalde zeldzame ziekten zouden kunnen tegengaan. Onze spreker gaf drie voorbeelden van aandoeningen waarvoor er al een gerichte behandeling bestaat.
De ziekte van Pompe is een metabole aandoening die wordt veroorzaakt door ophoping van glycogeen in de weefsels als gevolg van een tekort aan zuur alfa-1,4-glucosidase (lysosomaal enzym dat glycogeen hydrolyseert tot glucose). De ziekte wordt autosomaal recessief overgeërfd. Onbehandeld sterven kinderen met een infantiele vorm nog tijdens het eerste levensjaar aan hartfalen of ademhalingsinsufficiëntie als gevolg van een belangrijke linkerventrikelhypertrofie, spierzwakte of hypotonie. De volwassen vormen evolueren minder snel (er is nog een zekere enzymactiviteit) en worden gekenmerkt door progressieve spiervermoeidheid, waarbij ook de ademhalingsspieren aangetast kunnen worden. De aantasting van het hart uit zich in een hypertrofische cardiomyopathie die leidt tot hartfalen. Een verminderde zuurmaltaseactiviteit wijst op de ziekte van Pompe, en die diagnose kun je bevestigen met een genetisch onderzoek. Dankzij de aanwinsten in de moleculaire biologie is een recombinant humaan enzym ontwikkeld, dat sinds 2006 in de handel te verkrijgen is. De enzymsubstitutietherapie verbetert de levenskwaliteit en de levensverwachting van de patiënten en resulteert in een nagenoeg volledige regressie van het fenotype in de weefsels.
De ziekte van Fabry is een X-gebonden aandoening die veroorzaakt wordt door een tekort aan lysosomaal alfagalactosidase (GLA). Dat resulteert in een intracellulaire ophoping van sfingolipiden in tal van organen, waardoor die slecht gaan werken. De ziekte van Fabry is dan ook een 'systemische' ziekte. De klinische verschijnselen zijn vaak heterogeen en moeilijk te herkennen. Klassiek worden mannen meer aangetast, maar ook vrouwen kunnen verschijnselen vertonen (vaak later). Ook de ernst en de snelheid waarmee de symptomen verschijnen, is erg wisselend als gevolg van de heterogeniteit van de mogelijke mutaties en de wisselende residuele enzymactiviteit die daaruit voortvloeit. De eerste verschijnselen treden klassiek op bij jonge jongens in de vorm van pijn aan de extremiteiten, met soms vaatletsels op de romp of rond de navel. Bij jongvolwassenen zijn de nieren vaak aangetast, wat vaak leidt tot terminaal nierfalen en een sterke proteïnurie. Cardiovasculaire complicaties bepalen de prognose. De patiënten ontwikkelen een linkerventrikelhypertrofie, die ritmestoornissen veroorzaakt en/of kan leiden tot hartfalen. Het is interessant eraan te herinneren dat soms bij voorkeur de posterolaterale wand wordt aangetast. Dat geeft bij MRI een vrij kenmerkende late aankleuring. Het signaal van de ziekte van Fabry op 'T1-mapping' verschilt van dat van andere aandoeningen. T1-mapping leent zich dan ook zeer goed om ons op het spoor van de ziekte van Fabry te zetten. Niet zelden treedt cerebrovasculair lijden op (letsels van de witte stof). De oogaantasting wordt gekenmerkt door een cornea verticillata (zoals bij amiodarontoxiciteit). Bij mannen stoelt de diagnose op een bepaling van de enzymactiviteit van alfagalactosidase. Die diagnose wordt dan bevestigd door sequencing van het GLA-gen. Bij vrouwen is de enzymactiviteit vaak weinig veranderd (door de aanwezigheid van een compensatie die gebonden is aan het andere X-chromosoom). De diagnose berust dan in de eerste lijn op sequencing van het GLA-gen. Sinds het begin van de jaren tweeduizend beschikken we over een enzymsubstitutietherapie (tweemaandelijkse intraveneuze toediening van een recombinant enzym). Die verbetert de symptomen, beschermt het hart en de nieren en verbetert de prognose en de levenskwaliteit van de patiënten. Hoe sneller de behandeling gestart wordt, des te beter de resultaten. Vandaar het belang van een vroege diagnose. Onlangs is een geneesmiddel ontwikkeld dat het gebrekkige endogene enzym stabiliseert. Daarbij wordt gebruik gemaakt van de 'chaperonnetechniek'. Dat geneesmiddel kan enteraal toegediend worden. Gentherapie die gericht is tegen de betrokken mutatie of die de transcripts stabiliseert, heeft interessante resultaten opgeleverd bij muizen en wordt nu onderzocht bij de mens.
TTR-amyloïdose (autosomaal dominant) treedt op bij bejaarde patiënten of bij patiënten van middelbare leeftijd die een mutatie van het transthyretinegen vertonen. Transthyretine is een eiwit dat door de lever wordt gesynthetiseerd. Ophoping van amyloïdafzettingen in de weefsels veroorzaakt een disfunctie van verschillende organen (hoofdzakelijk de perifere zenuwen, de nieren en het hart). De hartaantasting kan ernstig zijn en wordt gekenmerkt door een linkerventrikelhypertrofie, die kan evolueren naar hartfalen en plotselinge dood. Het ecg toont klassiek microvoltages en een pseudo-infarctbeeld (niet altijd). Een MRI van het hart toont stoornissen bij de late aankleuring, meestal over de hele omtrek in het subendocardiale gebied. Diagnostisch interessant is dat een botscintigrafie met fosfaatmarkers tekent in het myocard, een teken dat zeer suggestief is voor TTR-amyloïdose.
Tot voor kort was de behandeling hoofdzakelijk ondersteunend (gaande tot hartof niertransplantatie) of causaal (levertransplantatie). Sinds kort beschikken we over geneesmiddelen die de ophoping van amyloïdafzettingen tegengaan. Die nieuwe geneesmiddelen interfereren met de stabiliteit en de oplosbaarheid van het gemuteerde TTR-eiwit (bijv. tafamidis) of verminderen de TTR-productie door interferentie met het boodschapper-RNA (bijv. patisiran en diflunisal). Momenteel lopen er tal van onderzoeken om de effecten van die geneesmiddelen op het hart te evalueren.
Aan de hand van die voorbeelden illustreerde prof. Linhart dat we dankzij een betere kennis van de pathofysiologische mechanismen van zeldzame ziektes nieuwe geneesmiddelen kunnen ontwikkelen en dat bij die patiënten tijdig een juiste diagnose gesteld moet worden, zodat ze maximaal baat kunnen vinden bij die behandelingen.
Referenties
- Laslett, L.J. et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol, 2012, 60 (25 Suppl): S1-49.
- Akinrinade, O. et al. Genetics and genotype- phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J, 2015, 36 (34), 2327-2337.
- Haas, J. et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J, 2015, 36 (18), 1123-1135a.
- van Rijsingen, I.A. et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol, 2012, 59 (5), 493-500.
- Ortiz-Genga, M.F. et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol, 2016, 68 (22), 2440-2451.
- Elliott et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J, 2014, 35 (39), 2733-2779.
- Moon, J.C. et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson- Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J, 2003, 24 (23), 2151-2155.
- White, J.A., Patel, M.R. The role of cardiovascular MRI in heart failure and the cardiomyopathies. Cardiol Clin, 2007, 25 (1), 71-95.
- Ugander, M. et al. Extracellular volume imaging by magnetic resonance imaging provides insights into overt and sub-clinical myocardial pathology. Eur Heart J, 2012, 33 (10), 1268-1278.
- Taylor, A.J. et al. T1 Mapping: Basic Techniques and Clinical Applications. JACC Cardiovasc Imaging, 2016, 9 (1), 67-81.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.