Definitie aorta-aandoeningen
In de overweging om genetische testing uit te voeren voor aorta-aandoeningen (AA) hebben we het in dit korte overzicht over aneurysmata en dissecties. Aorta aneurysmata gaan klassiek niet gepaard met symptomen en worden vastgesteld in het kader van (familiale) screening bij vermoeden van AA of bij beeldvorming voor andere indicaties. Om vast te stellen of een aortadiameter voor een bepaalde patiënt verhoogd is, dient men rekening te houden met het geslacht en de lichaamsbouw1. Hiermee kan men een z-score berekenen en als deze > 2 bedraagt, is er sprake van een verhoogde diameter. Aortadissecties worden onderverdeeld in type A en type B volgens de Stanford-classificatie. De prognose en behandeling van beide types zijn duidelijk verschillend, ten nadele van de type A dissecties.
De voornaamste onderliggende oorzaken van AA zijn hypertensie en atherosclerose. Er zijn evenwel ook patiënten zonder deze onderliggende risicofactoren die een verhoogd risico hebben en dan hebben we het over erfelijke vormen van AA - of Heritable Thoracic Aortic Disease (HTAD). Onder deze term vallen zowel gekende genetische vormen als familiale vormen, waarbij niet noodzakelijk een genetisch defect kan aangetoond worden. Het is in deze context dat genetische screening zeker moet overwogen worden, in de eerste plaats omdat dit kan helpen om personen met een risico op (fatale) aortadissectie tijdig op te sporen en te behandelen.
Bij wie dient genetische screening overwogen te worden?
Cardiologen en cardiochirurgen spelen een belangrijke rol bij het identificeren van personen die in aanmerking komen voor genetische screening. Bij vaststelling van aortadilatatie of -dissectie, en na uitsluiten van onderliggende hypertensie en/of belangrijke atherosclerose dienen patiënten klinisch geëvalueerd te worden ter exclusie van syndromale kenmerken (tabel 1) en dient een gedetailleerde stamboom met drie generaties te worden opgemaakt. Ook klinische evaluatie van eerste graadsverwanten kan belangrijk zijn, zeker in deze gevallen waar geen genetische oorzaak werd gevonden2, 3. Het identificeren van syndromale en/ of familiale vormen is belangrijk omdat hierbij de kans op het aantonen van een genetische oorzaak groter is. In dit verband heeft vroeg werk van Bart Loeys aangetoond dat de kans op het vinden van een genetische afwijking bij het marfansyndroom, de bekendste syndromale vorm, meer dan 90 % bedraagt4. Bij niet-syndromale vormen ligt dit percentage een stuk lager (tussen 15 en 20 %)5, 6, 7.
Meer recente populatiestudies tonen aan dat een positieve familiale anamnese voor aorta aneurysma/dissectie, ook na exclusie van het marfansyndroom, gepaard gaat met een sterk verhoogd risico op aneurysma/dissectie8, 9.
In geval van aortadissectie toont onderzoek ook bij sporadische gevallen (niet-familiaal/ niet-syndromaal) een verhoogde kans op het vinden van (pathogene) varianten en dit vooral in de jongere leeftijdsgroep10.
Welke genetische test en hoe?
Het tijdperk waarin genetische testing lang duurde en kostelijk was, ligt gelukkig achter ons. Met de huidige technieken is men in staat om uitgebreide gen-panels op een redelijke termijn en voor een aanvaardbare prijs uit te voeren. Dit wil evenwel niet zeggen dat uitgebreide panel testing of nog bredere 'whole exome of genome' testing bij iedereen geïndiceerd is. Omdat het aantal varianten met ongekende betekenis dat men aantreft evenredig toeneemt met het aantal geteste genen, blijft het ook nu nog altijd belangrijk om de genen die men voor een bepaalde aandoening kiest te onderzoeken, zorgvuldig te selecteren. In de setting van aortapathologie in associatie met lensluxatie kan men zeker overwegen om zich te beperken tot screening van het fibrilline-1 gen (FBN1). Gezien de belangrijke klinische overlap tussen verschillende syndromale vormen van HTAD, zoals het marfansyndroom en het loeys-dietzsyndroom, is het in deze gevallen raadzaam een set van genen te onderzoeken. Een vergelijkbare set kan aangewend worden voor testing in geval van niet-syndromale presentatie. Een lijst van zorgvuldig gescreende genen volgens een semiquantiatieve analyse voorzien door ClinGen werd gepubliceerd in 2018 en omvat volgende genen: ACTA2, COL3A1, FBN1, LOX, MYH11, MYLK, PRKG1, SMAD3, TGFB2, TGFBR1 en TGFBR211. Deze lijst is dynamisch en dient uiteraard regelmatig aangepast te worden op basis van meer recente gegevens. Bijkomende genen die in aanmerking komen zijn onder andere BGN, LTBP3, MAT2A, MFAP5, SMAD2, TGFB3 en THSD4.
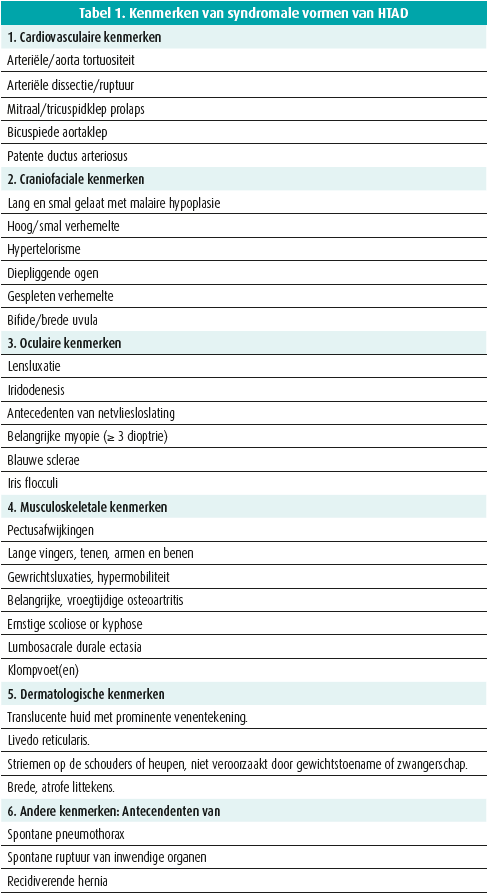
Genetische testing dient in ieder geval altijd gepaard te gaan met adequate counseling waarbij met patiënten en familieleden vooraf besproken wordt of er testing kan gebeuren, welke soort, en wat de consequenties hiervan zijn. De uitleg met interpretatie van de resultaten, eens die gekend zijn, dient opnieuw in een counseling setting te gebeuren waarbij in geval van een afwijkende variant duidelijke instructies voor verdere screening in de familie moeten gegeven worden. Een overzicht van de strategie voor genetische screening bij HTAD vindt u in figuur 1.
Waarom genetische testing uitvoeren bij HTAD?
Op een paar uitzonderingen na worden genetische vormen van HTAD autosomaal dominant overgeërfd, wat betekent dat er naar nakomelingen toe 50 % kans bestaat om de aandoening te hebben overgeërfd. In principe wordt de aandoening ook overgeërfd van een van beide ouders, hoewel men rekening moet houden met spontane mutaties in 25 % van de gevallen. Aantonen van een genetische oorzaak van aortapathologie laat dan ook in eerste instantie toe om familieleden van de patiënt in een vroeger stadium te identificeren om zo het risico op evolutie naar dissectie bij hen te reduceren. Daarnaast biedt dit ook mogelijkheden om bij een eventuele kinderwens stappen te zetten om transmissie te vermijden via pre-gestationele of prenatale testing.
Een bijkomend voordeel van kennis van de onderliggende genetische oorzaak is dat er nu al mogelijkheden zijn om een gen-specifiek beleid te voeren: in geval van pathologie veroorzaakt door genen betrokken in de TGFß pathway, die ook het loeys-dietzsyndroom veroorzaken, is het aangewezen om uitgebreide vasculaire beeldvorming (van hoofd tot klein bekken) te doen en wordt de grens voor profylactische heelkunde verminderd van 50 mm (voor het marfansyndroom - FBN1) naar 45 mm. Men kan nog meer in detail gaan en binnen één gen naar varianttypes kijken: varianten die leiden tot haplo-insufficiëntie (een verminderde kwantiteit) geven aanleiding tot een ernstiger aortafenotype dan varianten die aanleiding geven tot een dominant negatief effect (verminderde kwaliteit)12. Mogelijkheden voor een meer genspecifiek beleid zullen in de toekomst alleen maar toenemen - een mooie toepassing van precisiegeneeskunde.
Referenties
- Campens. L., Demulier. L., De Groote, K. et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am J Cardiol, 2014, 114 (6), 914-920.
- Thakker, P.D., Braverman, A.C. Cardiogenetics: genetic testing in the diagnosis and management of patients with aortic disease. Heart, 2020.
- Roman, M.J., De Backer, J. Hereditary thoracic aortic disease: How to save lives. [editorial]. J Thorac Cardiovasc Surg, 2021, 5223 (21).
- Loeys, B., De Backer, J., van Acker, P. et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat, 2004, 24 (2), 140-146.
- Campens, L., Callewaert, B., Muiño Mosquera, L. et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis, 2015, 10 (9).
- Arnaud, P., Hanna, N., Benarroch, L. et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Gen Med, 2019, 21 (9), 2015-2024.
- Regalado, E.S., Guo, D-c., Santos-Cortez, R.L.P. et al. Pathogenic FBN1 Variants in Familial Thoracic Aortic Aneurysms and Dissections. Clin Genet, 2016, 89 (6), 719-723.
- Chen, S.W., Kuo, C.F., Huang, Y.T. et al. Association of Family History With Incidence and Outcomes of Aortic Dissection. J Am Coll Cardiol, 2020, 76 (10), 1181-1192.
- Raunsø, J., Song, R.J., Vasan, R.S. et al. Familial Clustering of Aortic Size, Aneurysms, and Dissections in the Community. Circulation, 2020, 142 (10), 920-928.
- Guo, D-c., Hostetler, E.M., Fan, Y. et al. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J Am Coll Cardiol, 2017, 70 (21), 2728-2730.
- Renard, M., Francis, C., Ghosh, R. et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J Am Coll Cardiol, 2018, 72 (6), 605-615.
- Arnaud, P., Milleron, O., Hanna, N. et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patiënts with FBN1 pathogenic variants. Genet Med, 2021.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.