Définition des maladies aortiques
Dans le cadre de la réflexion sur la réalisa tion d'un dépistage génétique des maladies aortiques (MA), ce bref aperçu traitera des anévrismes et des dissections. Classiquement, les anévrismes aortiques ne s'accompagnent pas de symptômes, et sont diagnostiqués dans le cadre d'un dépistage (familial) en cas de suspicion de MA ou lors d'une imagerie réalisée pour d'autres indications. Pour déterminer si un diamètre aortique est augmenté chez un patient donné, il faut tenir compte du sexe et de la surface corporelle1. Ceci permet de calculer une z-score. Si celui-ci est > 2, le diamètre est augmenté. Les dissections aortiques sont subdivisées en type A et type B selon la classification de Stanford. Le pronostic et le traitement des deux types sont nettement différents, le type A étant le plus péjoratif.
Les principales causes sousjacentes des MA sont l'hypertension et l'athérosclérose. Cependant, certains patients ne présentant pas ces facteurs de risque sousjacents courent également un risque accru : il s'agit des formes héréditaires de MA - ou maladies héréditaires de l'aorte thoracique (HTAD). Ce terme recouvre à la fois les formes génétiques connues et les formes familiales lors desquelles on ne démontre pas nécessairement d'anomalie génétique. C'est dans ce contexte que le dépistage génétique doit assurément être envisagé, principalement parce qu'il peut aider à détecter et traiter à temps des personnes courant un risque de dissection aortique (fatale).
Chez qui faut-il envisager un dépistage génétique ?
Les cardiologues et les chirurgiens cardiaques jouent un rôle important dans l'identification des personnes entrant en ligne de compte pour un dépistage génétique. En cas de constatation d'une dilatation ou d'une dissection aortique, et après avoir exclu une hypertension sousjacente et/ou une athérosclérose importante, les patients doivent être évalués cliniquement afin d'exclure les caractéristiques syndromiques (tableau 1), et il faut établir un arbre généalogique détaillé sur 3 générations. L'évaluation clinique des membres de famille au premier degré peut également être importante, assurément dans les cas où une cause génétique n'a pas pu être trouvée2, 3. L'identification des formes syndromiques et/ou familiales est importante, car elle augmente la probabilité de démontrer une cause génétique. À cet égard, les premiers travaux de Bart Loeys ont montré que la probabilité de trouver une anomalie génétique en cas de syndrome de Marfan, la forme syndromique la plus connue, est supérieure à 90 %4. Dans les formes non syndromiques, ce pourcentage est beaucoup plus faible (entre 15 et 20 %)5, 6, 7.
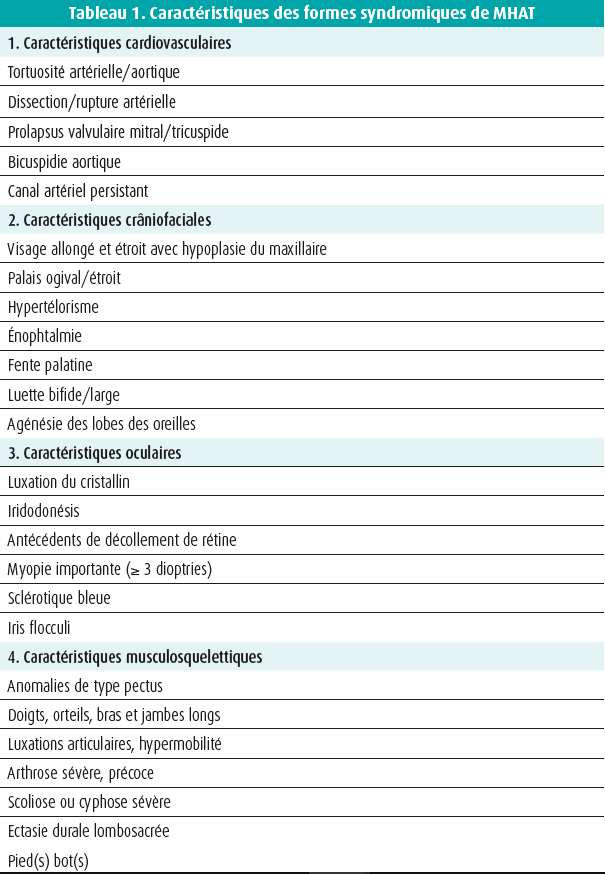

Des études de population plus récentes indiquent qu'une anamnèse familiale positive pour un anévrisme/une dissection aortique est associée à un risque très élevé d'anévrisme/de dissection, même si on a exclu un syndrome de Marfan8, 9.
En cas de dissection aortique, les études montrent également un risque accru de trouver des variants (pathogènes) dans les cas sporadiques (non familiaux/non syndromiques), et ce, surtout dans le groupe d'âge plus jeune10.
Quel test génétique utiliser et comment procéder ?
L'époque où les tests génétiques étaient longs et coûteux est heureusement révolue. Les techniques actuelles permettent de réaliser des panels de gènes étendus dans un délai et à un prix raisonnables. Pour autant, les tests avec un panel de gènes étendu ou un testing plus large de type séquençage panexomique ou pangénomique ne sont pas indiqués chez tout le monde. Comme le nombre de variants de signification inconnue détectés augmente proportionnellement au nombre de gènes testés, il reste important de sélectionner soigneusement les gènes que l'on choisit d'étudier pour une affection donnée. Dans le cadre d'une pathologie aortique associée à une luxation du cristallin, on peut assurément envisager de se limiter à l'analyse du gène de la fibrilline-1 (FBN1). Étant donné l'important chevauchement clinique entre différents syndromes de HTAD, tels que le syndrome de Marfan et le syndrome de Loeys-Dietz, il est conseillé, dans ces cas, d'étudier un set de gènes. Un set similaire peut être utilisé pour le testing en cas de présentation non syndromique. Une liste de gènes soigneusement sélectionnés selon une analyse semi-quantitative fournie par ClinGen a été publiée en 2018, et elle comprend les gènes suivants : ACTA2, COL3A1, FBN1, LOX, MYH11, MYLK, PRKG1, SMAD3, TGFB2, TGFBR1 et TGFBR211. Cette liste est dynamique et il va de soi qu'elle doit régulièrement être adaptée au moyen de données plus récentes. Entrent notamment en ligne de compte les gènes BGN, LTBP3, MAT2A, MFAP5, SMAD2, TGFB3 et THSD4.
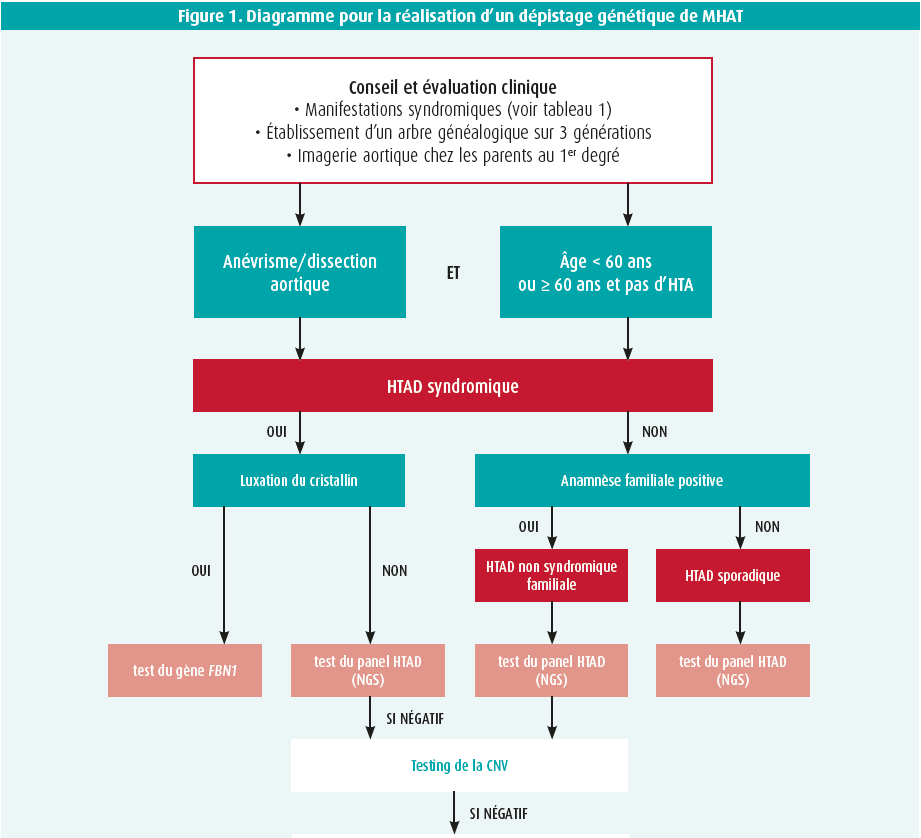
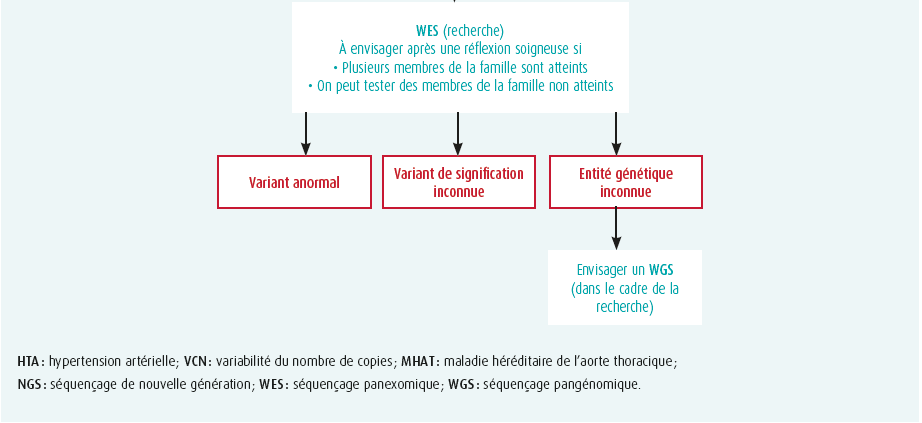
Tout testing génétique doit toujours s'accompagner d'un conseil adéquat, lors duquel on discute au préalable avec les patients et les membres de leur famille de la possibilité d'effectuer un test, du type de test et de ses conséquences éventuelles. Les explications et l'interprétation des résultats doivent à nouveau se faire dans le cadre d'une consultation. En cas de variant anormal, il convient d'apporter des consignes claires pour la poursuite du dépistage dans la famille. Un aperçu de la stratégie de dépistage génétique du HTAD peut être trouvé dans figure 1.
Pourquoi pratiquer un dépistage génétique en cas de MHAT ?
À quelques exceptions près, les formes génétiques de HTAD sont transmises sur un mode autosomique dominant, ce qui signifie qu'il y a 50 % de chances que les descendants aient hérité de l'affection. En principe, l'affection a également été transmise par un des deux parents, mais il faut tenir compte de mutations spontanées dans 25 % des cas. La mise en évidence d'une cause génétique d'une pathologie aortique permet donc en premier lieu d'identifier les membres de la famille du patient à un stade plus précoce, afin de réduire leur risque d'évolution vers une dissection. En outre, en cas de désir d'enfant éventuel, cette information permet de prendre des mesures pour éviter la transmission, via des tests prégestationnels ou prénataux.
Un autre avantage d'avoir connaissance de la cause génétique sous-jacente est qu'elle permet d'orienter le suivi : en cas de pathologie causée par des gènes impliqués dans la voie du TGFß, qui sont également à l'origine du syndrome de Loeys-Dietz, il est indiqué de procéder à une imagerie vasculaire étendue (de la tête au petit bassin), et la limite pour la chirurgie prophylactique est passée de 50 mm (pour le syndrome de Marfan - FBN1) à 45 mm. On peut encore aller plus loin et examiner les types de variants au sein d'un même gène : les variants qui entraînent une haploinsuffisance (réduction de la quantité) donnent lieu à un phénotype aortique plus sévère que les variants qui entraînent un effet négatif dominant (réduction de la qualité)12. À l'avenir, les possibilités d'une prise en charge plus spécifique des gènes seront toujours plus nombreuses - une belle application de la médecine de précision.
Références
- Campens. L., Demulier. L., De Groote, K. et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am J Cardiol, 2014, 114 (6), 914-920.
- Thakker, P.D., Braverman, A.C. Cardiogenetics: genetic testing in the diagnosis and management of patients with aortic disease. Heart, 2020.
- Roman, M.J., De Backer, J. Hereditary thoracic aortic disease: How to save lives. [editorial]. J Thorac Cardiovasc Surg, 2021, 5223 (21).
- Loeys, B., De Backer, J., van Acker, P. et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome.Hum Mutat, 2004, 24 (2), 140-146.
- Campens, L., Callewaert, B., Muiño Mosquera, L. et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis, 2015, 10 (9).
- Arnaud, P., Hanna, N., Benarroch, L. et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Gen Med, 2019, 21 (9), 2015-2024.
- Regalado, E.S., Guo, D-c., Santos-Cortez, R.L.P. et al. Pathogenic FBN1 Variants in Familial Thoracic Aortic Aneurysms and Dissections. Clin Genet, 2016, 89 (6), 719-723.
- Chen, S.W., Kuo, C.F., Huang, Y.T. et al. Association of Family History With Incidence and Outcomes of Aortic Dissection. J Am Coll Cardiol, 2020, 76 (10), 1181-1192.
- Raunsoslash;, J., Song, R.J., Vasan, R.S. et al. Familial Clustering of Aortic Size, Aneurysms, and Dissections in the Community. Circulation, 2020, 142 (10), 920-928.
- Guo, D-c., Hostetler, E.M., Fan, Y. et al. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J Am Coll Cardiol, 2017, 70 (21), 2728-2730.
- Renard, M., Francis, C., Ghosh, R. et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J Am Coll Cardiol, 2018, 72 (6), 605-615.
- Arnaud, P., Milleron, O., Hanna, N. et al. Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patiënts with FBN1 pathogenic variants. Genet Med, 2021.
Niets van de website mag gebruikt worden voor reproductie, aanpassing, verspreiding, verkoop, publicatie of commerciële doeleinden zonder voorafgaande schriftelijke toestemming van de uitgever. Het is ook verboden om deze informatie elektronisch op te slaan of te gebruiken voor onwettige doeleinden.